全基因组重测序是对已知基因组序列的物种进行不同个体的全基因组测序,并在此基础上对个体或群体进行差异性分析。基因组重测序方向是生物信息学中比较重要的一个分支,已经广泛应用到了农学、医学等领域,并对育种和靶向治疗起到了极具意义的指导作用。高通量测序技术出现后,重测序数据出现了指数型增长,海量数据的产生给生物信息学带来了极大的挑战。百迈客重测序分析平台为快速、高效、准确的完成数据分析,挖掘数据中的科学意义,提供了一条捷径。其常规分析内容包括:测序数据质量评估,序列比对,SNP、InDel、SV、CNV等变异检测和注释,变异所在基因的功能注释等;在此基础上还可以进行深入挖掘,包括:SNP分析、InDel分析、突变基因分析、Circos制图、基因引物设计。
重测序,即重新测序,是对已有参考基因组的物种进行个体或群体的基因组测序,利用高性能计算平台和生物信息学方法,在全基因组水平扫描变异位点(SNP、InDel、SV、CNV),快速准确的定位差异基因,并可应用于群体遗传学研究、关联分析、进化分析等。
目前重测序技术已广泛应用于农学、医学等各个研究领域,包括性状相关候选基因筛选、动植物育种、单基因病筛查、癌症筛查等,快速准确,对育种和临床诊断有很好的指导作用。

基本分析:只需要选择原始测序数据和参考基因组,一键提交即可完成常规的重测序分析内容,包括:测序数据质量评估,序列比对,SNP、InDel、SV、CNV等变异检测和注释,变异所在基因的功能注释等。流程分析用时因参考基因组大小、样品数目、测序数据量等而异,200M的参考基因组,4个样品,测序深度平均为20X(4G/样品),分析耗时约24h。
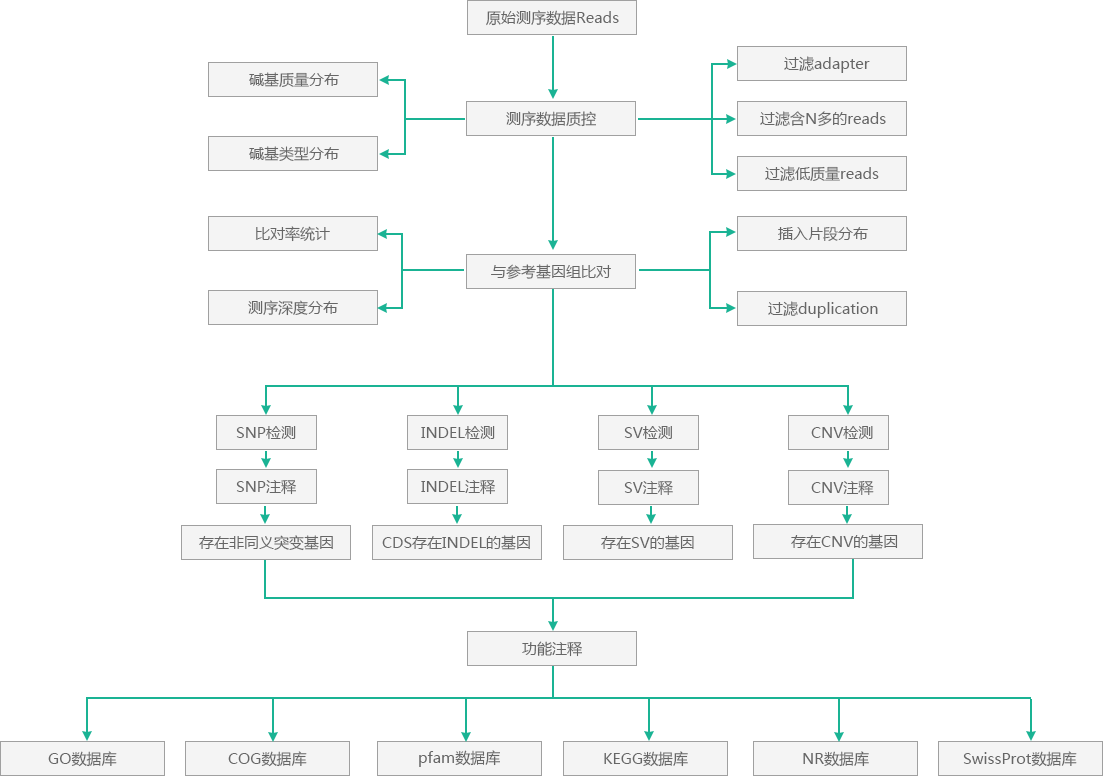
提取样本DNA后进行建库和高通量测序得到该样本的基因组序列数据,选择合适的参考序列进行重测序数据分析,分析流程如下。
SNP和InDel的检测主要使用GATK[1]软件包实现。根据测序序列在参考基因组的定位结果,使用Picard进行去重复(Mark Duplicates)、GATK进行局部重比对(Local Realignment)、碱基质量值校正(Base Recalibration)等预处理,以保证检测得到的SNP准确性,再使用GATK进行单核苷酸多态性(Single Nucleotide Polymorphism,SNP)的检测,过滤,并得到最终的变异位点集。
具体检测过程如下:
(1) 用BWA[2]进行序列比对得到的比对结果,使用Picard的Mark Duplicate工具去除重复,屏蔽PCR duplication的影响;
(2) 使用GATK进行InDel Realignment,即对存在插入缺失比对结果附近的位点进行局部重新比对,校正由于插入缺失引起的比对结果错误;
(3) 使用GATK进行碱基质量值再校准(Base Recalibration),对碱基的质量值进行校正;
(4) 使用GATK进行变异检测(Variant Calling),主要包括SNP和InDel;
(5) 使用GATK对得到的变异结果进行校正,选取可靠的变异结果。
用SnpEff[3]对变异(SNP、Small InDel)进行注释和功能影响预测。根据变异位点在参考基因组上的位置以及参考基因组上的基因位置信息,可以得到变异位点在基因组发生的区域(基因间区、基因区或CDS区等),以及变异产生的影响(同义非同义突变等)。
SV(结构变异)指基因组水平上大片段的插入、缺失、倒置、易位等。结构变异使用BreakDancer[4]检测。BreakDancer首先基于序列与参考基因组的比对结果,得到测序数据文库的插入片段大小和方差,然后通过查找序列和参考基因组之间的异常比对结果(插入片段发生偏离、比对方向不一致等),寻找可能的结构变异。
Identifying the Genome-Wide Sequence Variations and Developing New Molecular Markers for Genetics Research by Re-Sequencing a Landrace Cultivar of Foxtail Millet
研究背景
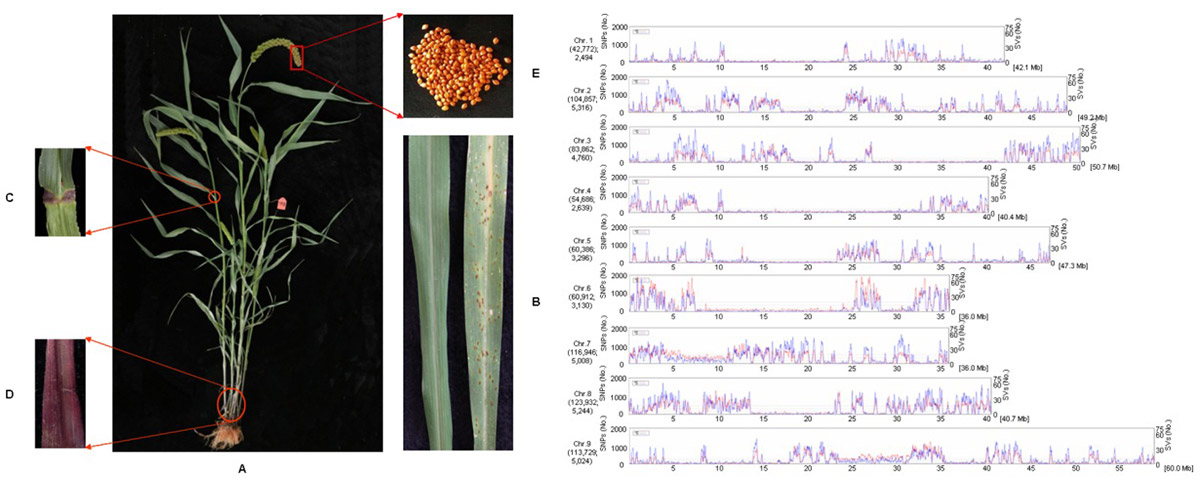
由于良好的抗旱抗逆特征,谷子开始作为模式作物被广泛研究。不同品种的谷子性状差异主要是由基因差异造成的,本研究通过个体重测序技术横向比较谷子Shi-Li-Xiang品种(SLX)与Yugu1品种和Zhanggu品种的基因组差异,挖掘基因标记用于全基因组分型和辅助育种。
实验设计
提取三个品种的水稻DNA,采用Illumina测序平台进行高通量测序,每个样品基因组覆盖度达 93%,平均测序深度为 11X,之后进行生物信息学分析。
信息分析
与 Yugu1 基因组比较,SLX 品种鉴定出 762,082 个 SNPs,26,802 个 InDels,插入或缺失片段大小在 1-5bp,10,109 个 SVs。与 Zhanggu 基因组比较,SLX 品种鉴定出 913,454 个 SNPs,28,546 个 InDels 和 12,968 个 SVs。发现40%的SNP存在于NB-ARC结构域、蛋白激酶或富含亮氨酸的重复序列中。非同义突变与同义突变的SNP比例很高,与水稻、高粱等物种一致,说明植物抗病蛋白的分化也许是由病原菌的进化压力导致的。
在SLX和Yugu1两个品种中,具有多态性的标记中有465个SNPs和146SVs准确性在90%以上,可以作为DNA标记进行全基因组基因分型和分子标记辅助育种。