LncRNA是非编码RNA的重要组成部分。近年来,随着测序以及其他分析技术的发展,大量lncRNA已经被鉴定,并且越来越多的证据表明lncRNAs参与多种生物过程,并且发挥关键作用。因此,lncRNA的分析也就越来越受到大家的重视,且将lncRNA的分析应用到众多新的测序物种中,呈现多元化发展的迹象。另外,lncRNA在许多新的领域发挥着重要作用,如在细胞分化和个体发育中的研究愈发成熟;在研究lncRNA与人类疾病的关联上的案例也越来越多,尤其是在癌症研究领域,lncRNA可能是治疗癌症的突破口之一;近年的研究还发现在植物生长、发育和环境胁迫适应等过程中lncRNA也发挥着重要的调控作用。国内lncRNA研究也逐渐火热起来,前不久出炉的2015年NSFC资助结果表明lncRNA项目较去年翻一番,不出所料又一研究新贵诞生,那就是Lnc RNA,其项目多达300项。
随着lncRNA研究的深入,科研工作者需要投入的时间和精力也越来越大,研究经费也越来越多,百迈客云平台为了给更多的科研工作者解决这些困扰,特别开发了一个lncRNA分析平台,给科研工作者提供了一个能快速研究lncRNA的平台,给用户搭建了一座快速研究lncRNA的桥梁,给科研工作者提供了一个快速通往lncRNA的机会。
长链非编码RNA与细胞分化和个体发育:
长链非编码 RNA 的表达不仅具有细胞型和组织特异性, 一些长链非编码 RNA 还仅在真核生物发育过程的特定阶段表达。对线虫和果蝇发育过程中长链非编码 RNA 的表达研究发现, 这类 RNA 分子呈现出动态的表达变化, 多数长链非编码RNA 具有精确的时间和空间表达模式, 有的表达模式还保守地存在不同种的果蝇中。
长链非编码RNA与人类疾病:
研究发现, 长链非编码 RNA 的表达或功能异常与人类疾病的发生密切相关, 其中就包括癌症、退行性神经疾病在内的多种严重危害人类健康的重大疾病, 具体表现为长链非编码 RNA 在序列和空间结构上的异常、表达水平的异常、与结合蛋白相互作用的异常等。
长链非编码RNA与植物生长发育、春化作用、非生物环境胁迫适应性的调控作用:
在自然界中许多高等植物需要通过冬季的低温阶段实现从营养生长到生殖生长的时期转化,这一生物学过程称作春化作用。小麦(Triticum aestivum L.)和油菜(Brassica napus L.)等作物以种子为产品器官,生产上往往通过茬口安排和栽培措施使植株尽早通过春化作用,以促进花芽形成和花器官发育,而大白菜(B rapa ssp.pekinenesis)和甘蓝(B.oleracea)等作物以叶球等营养器官作为产品器官,生产上则设法避免低温引起的春化作用,以保证产品器官的充分生长。FLOWERING LOCUS C(FLC)作为一种重要的开花抑制蛋白负调控春化作用,参与植株从营养生长向生殖生长的转化过程。在植物中很多功能的调控都离不开lncRNA参与。
长链非编码RNA测序分析平台可以进行标准分析和个性化分析,其中标准分析包括:差异表达基因分析、基因结构分析、新lncRNA预测及靶基因预测等。设定参数后点击提交进行分析,分析完成后在流程定制页面下生成标准化结题报告,实现一键式生成。
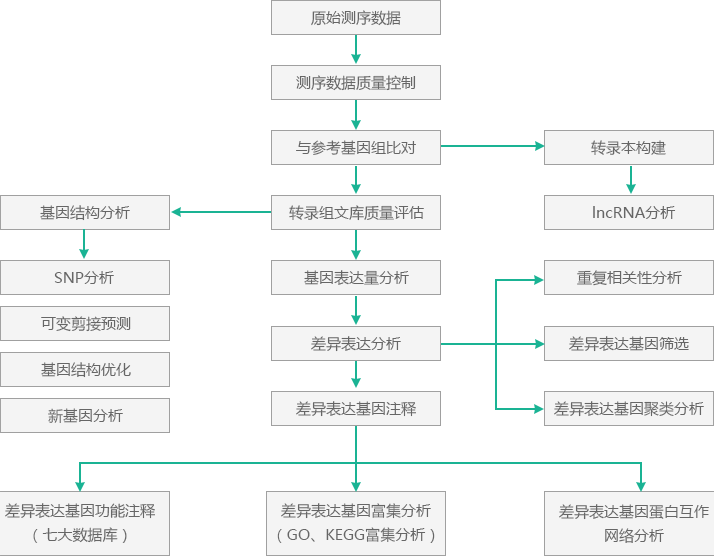
对Raw Data进行数据过滤,去除其中的接头序列及低质量Reads获得高质量的Clean Data。将Clean Data与指定的参考基因组进行序列比对,获得Mapped Data。基于Mapped Data,进行插入片段长度检验、随机性检验等测序文库质量评估;进行表达量分析、可变剪接分析、新基因发掘和基因结构优化等。根据基因在不同样品或不同样品组中的表达量进行差异表达分析、差异表达基因功能注释和功能富集等高级分析。
获得Clean Reads后,将其与参考基因组进行序列比对,获取在参考基因组或基因上的位置信息,以及测序样品特有的序列特征信息。
TopHat2[1]是一个高效的序列比对软件。它以高通量Reads比对软件Bowtie[2]为基础,将长链非编码测序Reads比对到基因组上,然后通过分析比对结果识别外显子之间的剪接点(Splicing Junction)。这不仅为可变剪接分析提供了数据基础,还能够使更多的Reads比对到参考基因组,提高了测序数据的利用率。
长链非编码测序数据中,只有比对到参考基因组上的数据才能用于后续分析。因此,将比对到指定的参考基因组上的Reads称为Mapped Reads,对应的数据称为Mapped Data。
新lncRNA的预测包含基本筛选和潜在编码能力筛选两部分。基本筛选保留的转录本,去除掉具有潜在编码能力的转录本,余下的转录本即为新预测的lncRNA。基本筛选主要由4个部分组成:
(1)去除基因组数据库中的mRNA(转录本及其剪接体);
(2)选择长度≥200bp,Exon个数≥2的转录本;利用cufflinks计算每个转录本的FPKM,选择FPKM≥1.5的转录本;
(3)同时被两个拼接软件拼接得到的lncRNA也筛选出来,得到最终的新预测lncRNA集进行后续分析;
(4)利用cuffcompare信息筛选lincRNA,intronic lncRNA, anti-sense lncRNA等不同类型的lncRNA。
长链非编码RNA按照其来源可分为Antisense lncRNA (反义长非编码RNA),Intronic transcript (内含子非编码RNA),Large intergenc noncoding RNA(lincRNA),Promoter-associated lncRNA(启动子相关lncRNA),UTR associated lncRNA(非翻译区lncRNA)五种类型。
最后再根据编码能力预测软件预测新lncRNA的编码能力,是否具有编码潜能是判断转录本是否为lncRNA的关键条件,综合研究中应用最广泛的编码潜能分析方法进行lncRNA筛选,主要包括:CPC分析、CNCI分析、pfam蛋白结构域分析三种方法。
CPC[3] (Coding Potential Calculator)分析是一种基于序列比对的蛋白质编码潜能计算工具。通过转录本与已知蛋白数据库比对,根据转录本各个编码框的生物学序列特征评估其编码潜能。
CNCI[4](Coding-Non-Coding Index)分析是一种通过相邻核苷酸三联体特征区分编码-非编码转录本的方法,该工具不依赖于已知的注释文件。
Pfam[5] 数据库是最全面的蛋白结构域注释的分类系统。蛋白质是由一个或多个结构域组成的,而每个特定结构域的蛋白序列具有一定保守性。Pfam将蛋白质的结构域分为不同的蛋白家族,通过蛋白序列的比对建立了每个家族的氨基酸序列的HMM统计模型。能比对上的转录本即为具有某个蛋白结构域的转录本,即认为具有编码能力,而无比对结果的转录本是潜在的lncRNA。
结合以上3种分析结果,对所有鉴定得到的潜在的lncRNA转录本进行统计。将3种分析结果取交集,用于后续lncRNA。
Genome-wide discovery and characterization of maize long non-coding RNAs [1].
研究背景
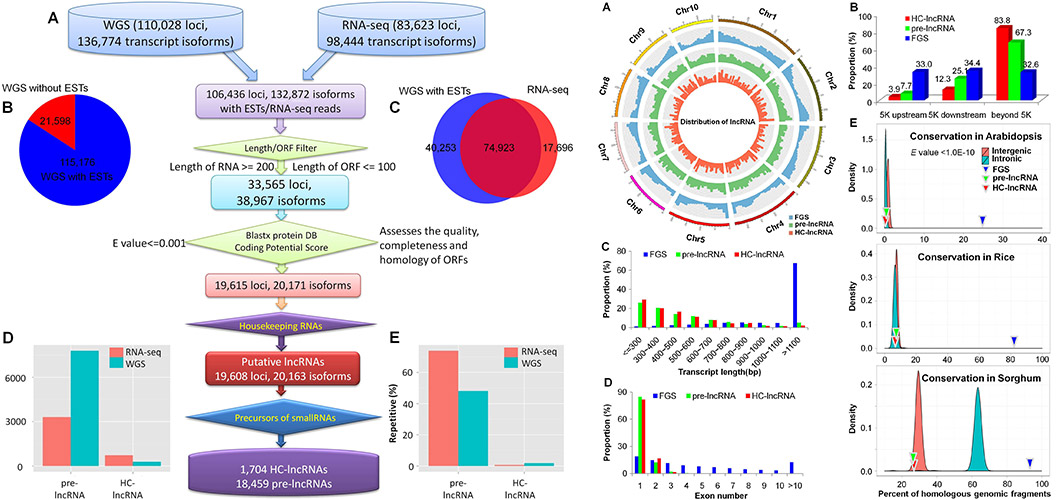
使用生物信息学对玉米全长cDNA测序预测出1802条lncRNA,其中60%可能是小RNA的前体。调查发现,相比人类和其他哺乳动物,植物中大量的lncRNA往往表现出较低组织特异性,并作为天然miRNA靶标、染色质修饰或蛋白分子的重新定位。为了预测一个更全面更多的玉米lncRNA,我们综合了maize-b73参考基因型的各种信息,如公开发表EST库、玉米全基因组测序的注释和30种不同实验和发展阶段的RNA测序数据集。
研究概况
本研究利用基因组测序和30 组不同转录组测序数据对玉米的lncRNA 进行分析,共鉴定得到20,163 个lncRNA,其中18,459 个位点是pre-lncRNA,1,704 个HC-lncRNA。7% 的lncRNA 与编码的mRNA 序列重叠,81% 的lncRNA仅具有一个外显子。54% 的lncRNA 仅在一个组织中检测到,仅有8% 的基因在一个组织中检测到,说明lncRNA 的表达具有很高的组织特异性。