

从狭义上来讲,转录组是指能转录出来的所有mRNA的总和,而从广义上来讲是指细胞内所有转录产物的集合(包含mRNA和非编码RNA)。miRNA, circRNA以及lncRNA作为一类具有调控功能的非编码RNA被广泛的研究,并且调控对象均与mRNA相关。而全转录组测序所研究的对象,即是特定细胞在某一时间所转录出的是编码(mRNA)和非编码RNA(包含lncRNA, circRNA, miRNA)。在生物体内,任何一类RNA分子都不是孤立的,他们行使不同的生物学功能,参与调控机体的重要通路。因此,多种RNA的整合研究势在必行。
目前,单一的编码或非编码RNA研究已经不能满足科研工作者对复杂生命体的机制的探索,整合多种RNA并从不同角度的联合分析进而研究其潜在的调控机制已经成为解释生物学现象必不可少的利器!
随着测序成本的不断降低及通量的不断提高,也使得同时分析一份样品的mRNA, lncRNA, circRNA和miRNA在技术上和经济上成为可能。可以全面的揭示生物体复杂的调控机制,进而广泛的应用于动植物的生长发育和凋亡、免疫调控、疾病发生发展以及药物靶标研究等。

全转录组联合分析平台基本分析流程可以全局的展示多种RNA以及差异情况,对4种RNA之间进行共表达分析,ceRNA网络构建,关键RNA筛选以及关键mRNA的KEGG整合通路网络分析。设定参数后点击提交进行分析,分析完成后在流程定制页面下生成标准化结题报告,实现一键式生成。
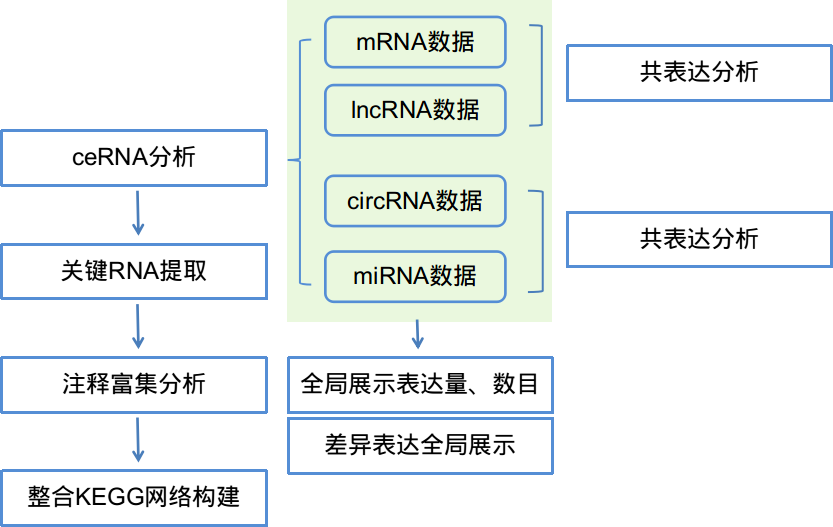
对二代测序数据,首先使用百迈客自主研发的流程对各种RNA单独进行分析(包含lncRNA, mRNA, circRNA以及miRNA分析流程),进而采用全转录组联合分析平台首先采用自主研发的程序进行多种RNA全局整合以及差异RNA的整合。
对差异RNA采用皮尔森相关性分析对样品的表达量进行公表达分析[1]。基于RNA(mRNA, lncRNA以及circRNA)与miRNA的靶向关系进行ceRNA网络的构建。所有RNA与其他类型的RNA两两之间形成候选的关系对,根据其与miRNA结合的情况,进行超几何分布统计分析,对所有关系对的统计结果进行FDR校验获得ceRNA关系对[2]。从这些背景的ceRNA关系对种获取差异RNA的一步近邻网络。采用随机游走算法选择一步近邻网络中重要的节点。
基于上述信息,对重要的节点进行功能富集分析以及整合的KEGG网络分析,进而找到感兴趣的重要的基因及其所调控的基因。
研究背景
越来越多迹象表明非编码RNAs可以通过神秘的机制来调控基因表达。就长度和结构而言,ncRNA超家族包含小RNA(miRNA),长非编码RNA(lncRNA),以及新定义的环状RNA(circRNA)。事实上,RNA在机体中起着重要的作用。作者通过对10个成人腰椎NP组织相同的RNA样品的mRNA, miRNA, circRNA, lncRNA的分析,展示了人类腰椎键盘的RNA的风貌并在RNA的互作机制提出了新的见解。
实验设计
研究选用5个正常尸体捐献者以及5个退变性的腰椎间盘(NP)样品,采用lncRNA芯片,microRNA芯片(miRCURYTMLNA Array)以及circRNA芯片进行数据分析。
生物信息分析
对芯片数据经处理后获得每种RNA的表达情况,采用层次聚类以及箱式图对不同样品的不同RNA的表达量进行全局展示(图1)。同时发现染色体1以最高的水平转录RNA,然而染色体Y是最低的(图2)。采用TargetScan 以及miRanda预测miRNA的靶点。在circRNA和miRNA中发现了5310个结合位点。从前20个差异的miRNA以及前20个差异的circRNA中,找到了7个失调的miRNA可能通过84个潜在的结合位点与32个circRNA互作,然而20个失调的circRNA可能通过143个潜在的结合位点与99个miRNA结合。这些核心的互作可能与免疫特权器官组织紧密相连。
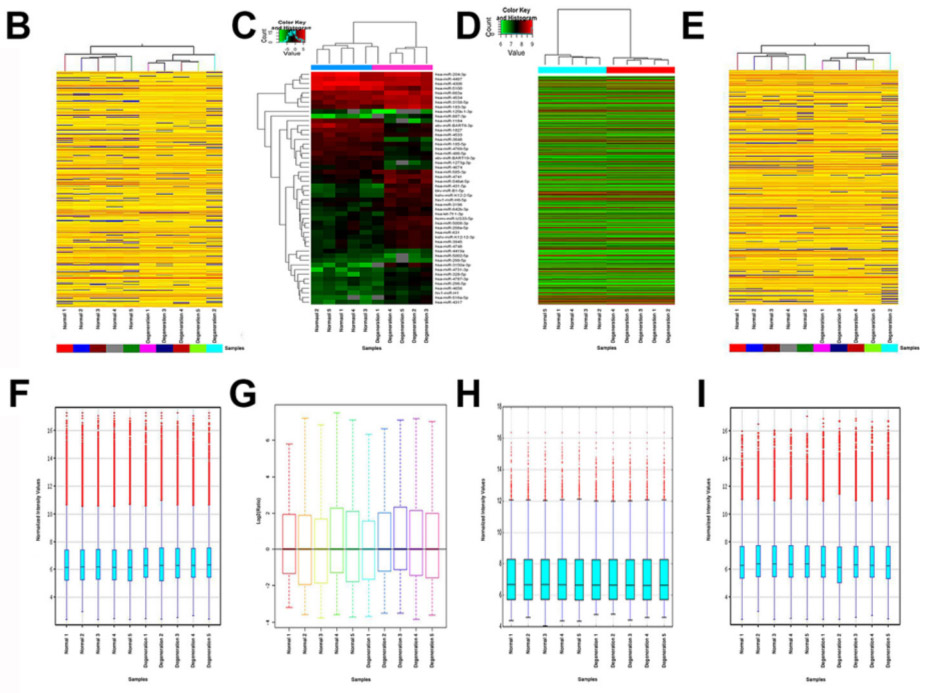
图1. 不同RNA表达的聚类热图和箱式图
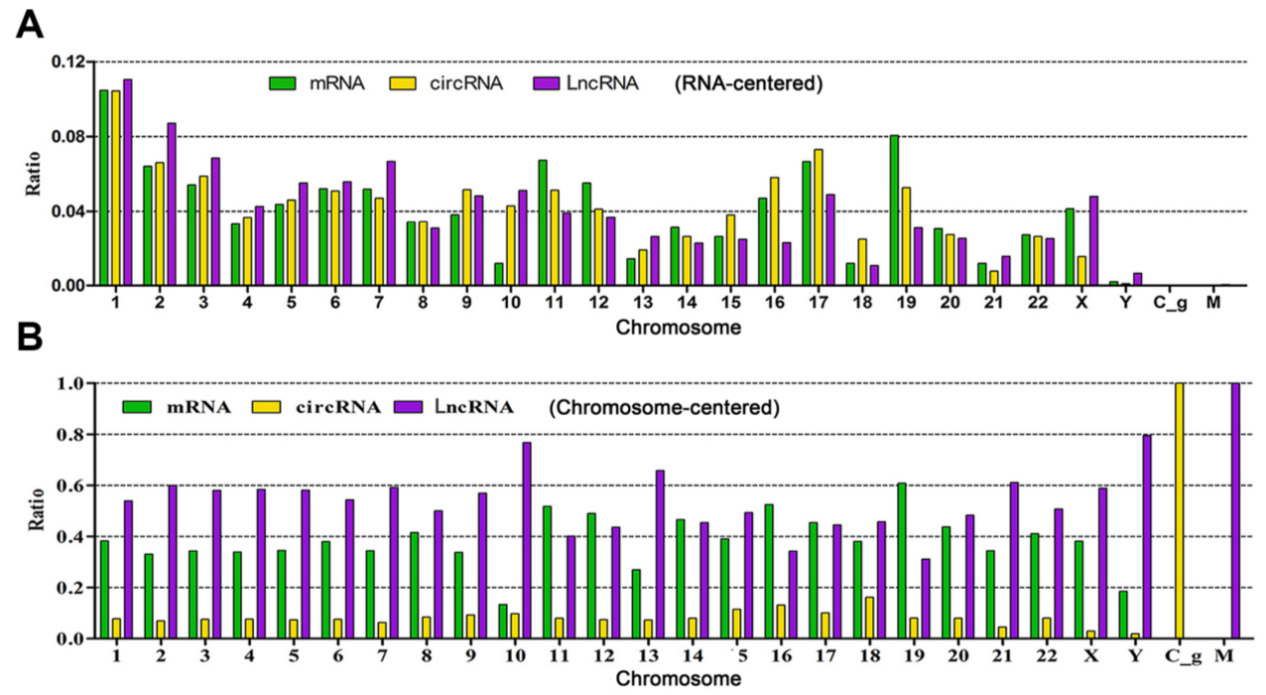
图2. 不同RNA在不同染色体中的相对表达量