


百迈客云平台微生物多样性分析平台是一款结合多年微生物多样性项目分析经验开发的包含一键式标准化基本分析和个性化多样性分析的集成式分析平台:基本分析涵盖了目前微生物研究的主流分析内容,分析内容丰富全面,分析结果以结题报告的形式给出;
个性化分析内容多样,可以根据基本分析报告及研究目的选取样本、灵活设置参数,实现个性化需求;Windows操作界面、简单快捷、即买即用。
微生物是地球上已知种类最多、数量最大、分布最广的生物类群。它对环境敏感,反应迅速与环境和生命体密不可分。研究环境,进化,生物体,疾病,和历史等问题,研究相关(环境)微生物往往更能说明问题。微生物多样性研究采用宏基因组测序方法,以特定生境中的整个微生物群落作为研究的对象,不需对微生物进行分离培养,而是提取环境微生物总DNA 进行研究。摆脱了传统研究中微生物分离培养的技术限制,采用新一代高通量测序技术对环境微生物样品的DNA直接测序。能满足常规微生物群体的物种分类研究、群落结构、系统进化、基因功能分析以及物种间的代谢网络等分析。
目前微生物研究已广泛应用于工农业、医学、生物能源、环境治理、生物技术等方面。
微生物多样性分析平台可以进行标准分析和个性化分析,其中标准分析包括:物种分类分析(包括:群落结构分析、物种Heatmap、物种系统进化树)、单样品多样性分析(α- 多样性指数、稀释曲线、Rank-Abundance 曲线)、多样品多样性分析(UniFrac分析、样品Heatmap、PCA/PCoA分析、RDA/CCA 分析、样品层次聚类)、组间样品差异分析(Venn 图、LefSe分析、Metastats分析)等。设定参数后点击提交,一键式生成标准化结题报告。
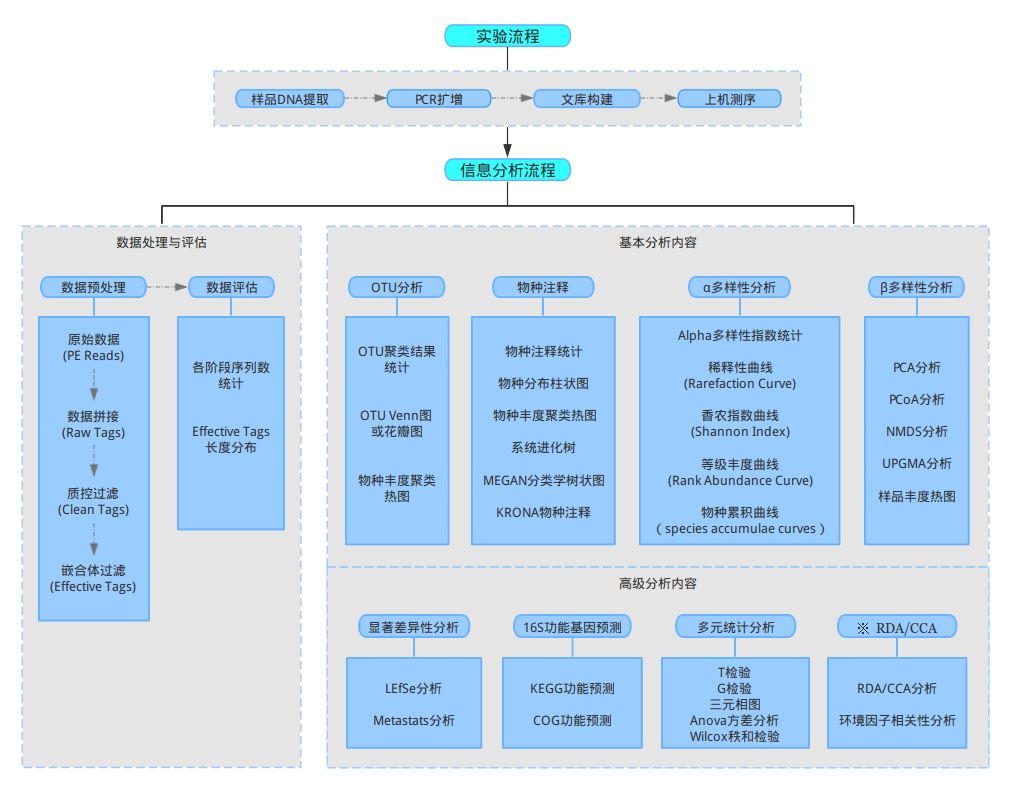
微生物多样性分析是基于OTU (分类操作单元)进行的,在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。一般情况下,如果序列之间的相似性高于97% 就可以把它定义为一个 OTU。
以24个16s rDNA样品为例,靶标区域V4区,Miseq测序,技术策略如下:
1.PCR扩增:为了降低测序建库成本,我们在扩增V4区时在引物序列上加上一段Index(barcode)序列,然后分别进行V4区的扩增,这样不同样品得到的PCR产物会包含不同的Index序列,可用于后续分析时分样;
2.混样:把(1)中的24个PCR产物混合到一起,但是为了保证测序时不同样品中reads数分布尽量均匀,我们要以等量的PCR产物进行混合;
3.建库及测序:按照Illumina公司提供的标准流程进行建库,然后上机测序,选择PE 2X150bp(paired end)测序模式,见图3;
4.PE数据合并:V4区254bp,从每一端我们测得150bp,则两端测到的序列会有46bp的重叠,我们可以根据对应序列的重叠部分将两条序列合并为一条,然后进行后续的生物信息学分析;
5.OTU 聚类:QIIME[1](version 1.8.0, http://qiime.org/index.html)软件中的 UCLUST[2] (version 1.2.22,http://www.drive5.com/uclust/downloads1_2_22q.html);
6.物种分类分析,单样品微生物多样性分析,多样品多样性分析。
Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest[1]
研究背景
叶片上的微生物群落结构与植物机体的进化发展、植物体获取生态资源的功能特性研究以及植物生长/死亡息息相关,目前已知了大量的与宿主植物体生长、死亡以及功能相关的多个细菌类群,本研究旨在量化引起植物相关微生物多样性变化的驱动因素。
实验设计
本研究选取巴拿马地区热带森林的57 个树品种的叶际,每种树采1-5 个样品,采用16S rDNA测序。共检测到7,293 个OTU,每个样本平均为418 个,稀释曲线证明没有测完全,预估OTU 数为11,615,发现有104 个核心叶际微生物OTU。
信息分析
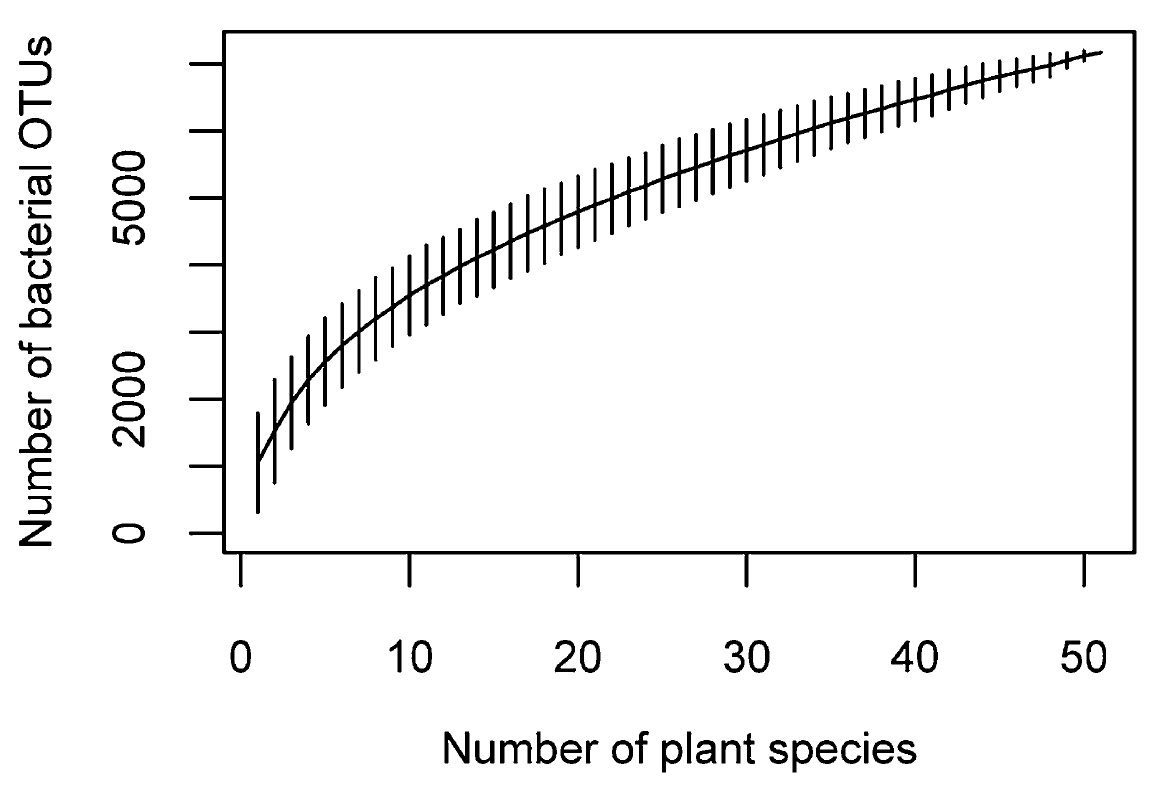
图1Collector’s curve(植物种类-微生物OTU浓度曲线):反映了随宿主种类增多,所含的微生物OTU数量关系变化情况。该图显示了OTU数量只在一定范围内受采样的宿主植物影响较大,随着采样植物种类增加,影响越来越小。
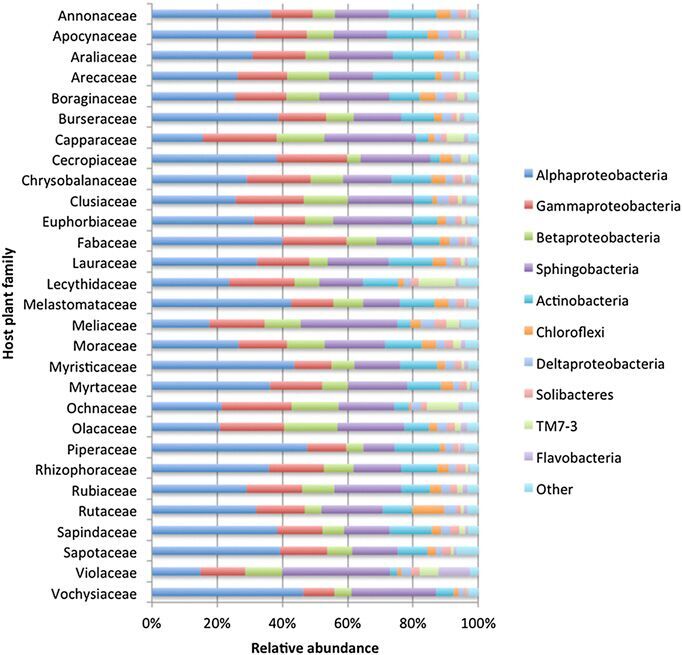
图2.分类热图:各类宿主植物包含的微生物分类丰度.反映了不同种类植物叶际所含有的微生物群分类;该图显示出普遍、大量存在于森林中几乎所有植物叶际的微生物分类——“叶际核心微生物”,该核心物种存在于超过95%的植物叶片上,含有104个细菌OTU,分布于8个门34个属,能够代表超过73%的细菌序列差异。
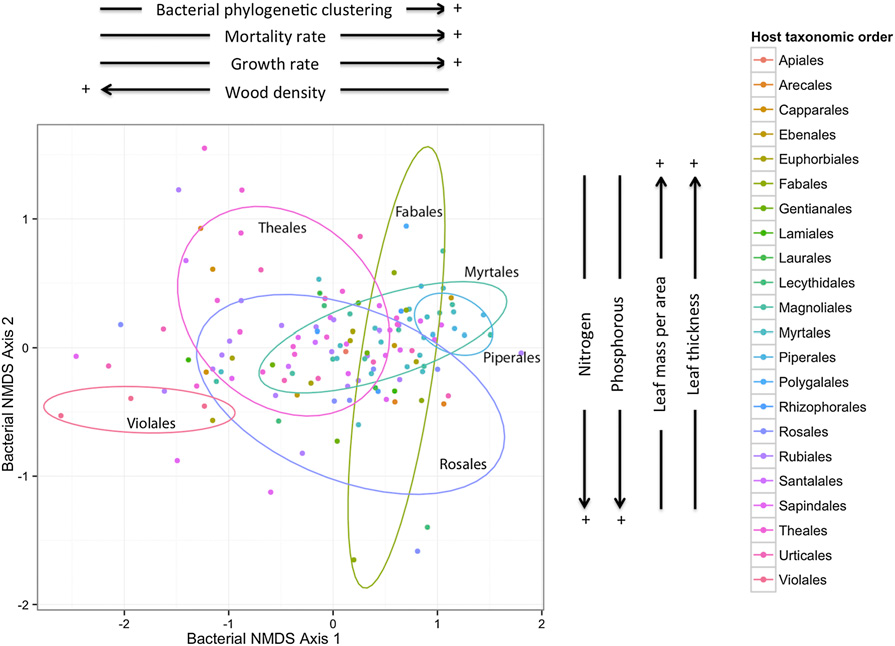
图3.NMDS主成分分析图:反应叶际细菌群落分类变化的驱动力.图中是宿主植物的两组特性:第一组(横坐标)是对于微生物群落变化最主要(P<0.05)特性,包括树林密度、生长率和死亡率;第二组(纵坐标)特性是与微生物是影响微生物群落分类的最主要(P<0.05)特性,包括单位面积内的叶片重量、叶片厚度以及叶片的氮磷含量。图上反映出:叶际微生物聚集属于系统发生,并且使得OTU之间联系越来越紧密。系统发生聚集的数量级与主成分1(树林密度、生长率和死亡率)密切相关。
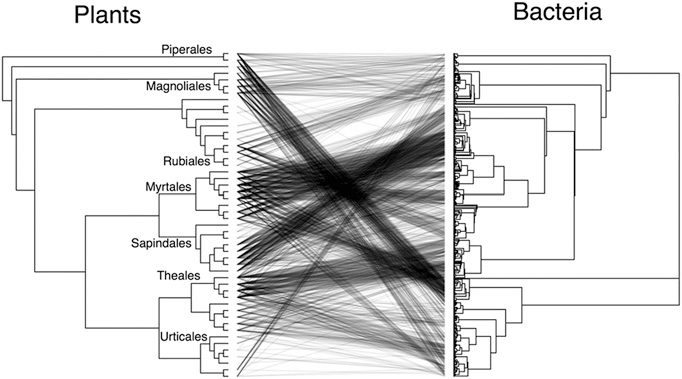
图4.系统发育树关系图:通过测试两者总体的进化联系和鉴别个体的“宿主-微生物联系”比随机的“宿主-寄主关联测试”更强,从而量化宿主种类与寄主OTU之间进化关联的强度图中显示了宿主植物种类与微生物OTU总体进化关系(P <0.001)和分支进化关联(P <0.05)。
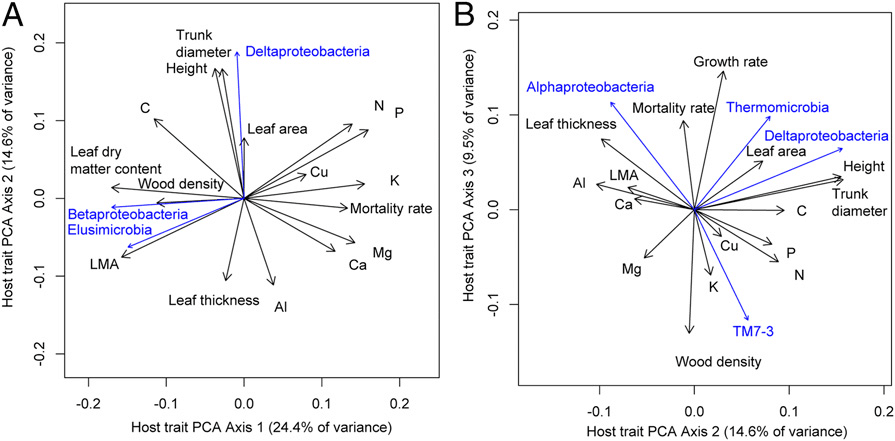
图5.系统发育树关系图:微生物群落结构与宿主植物特有功能间的关系.图中显示了Barro Colorado Island(A)和Panama(B)两个岛上217个宿主植物种类的三个主要特性(axis 1,axis 2,axis 3)的主成分分析结果。其中axis 1(24.4% of variance),axis 2(14.6% of variance),axis3(9.5% of variance)。这些轴线解释了49%的数据变化。箭头方向指向关联特性,箭头长度表明关联强度;黑箭头表示植物相关特性,蓝箭头表示两个主成分间有重要关联(P < 0.05)。
分析结果
信息分析表明:影响微生物群落组成变异影响最大的两个因素,一是树木的密度以及生长死亡率,二是叶片厚度及氮磷含量。PCA 分析发现三种主要的影响微生物多样性的因素为叶片干物质含量及营养含量,植株成熟期的直径及高度,木质密度生长率以及死亡率。