

百迈客云平台小RNA测序分析平台是结合百迈客多年sRNA测序项目分析经验开发的一款包含一键式标准化分析流程和多样性个性化分析流程的集成式分析平台。标准分析丰富全面以结题报告的形式给出。个性化分析内容多样,可以根据研究目的灵活设置参数,实现个性化需求。接近windows的操作界面、操作简单,即买即用。
Small RNA是一种小的非编码RNA,长度为18-30个核苷酸,几乎存在于所有的生物体中,包括:miRNA、siRNA和piRNA。Small RNA通过多种多样的作用途径,包括mRNA降解、翻译抑制、异染色质形成以及DNA去除,来调控生物体的生长发育和疾病发生,在人、动植物的转录和转录后调控过程中起着重要的作用。
Small RNA高通量测序分析目前被广泛应用于动植物生长、分化、发育和凋亡等发育调控研究;药物、胁迫等环境适应性研究;病毒、真菌、细菌等免疫互作研究等方面。
小RNA测序分析平台可以进行标准分析和个性化分析,其中标准分析包括:miRNA的鉴定与预测;miRNA表达量分析;miRNA靶基因预测;miRNA靶基因注释分类及富集等。设定参数后点击提交进行分析,分析完成后在基本分析页面下生成标准化结题报告,实现一键式生成。
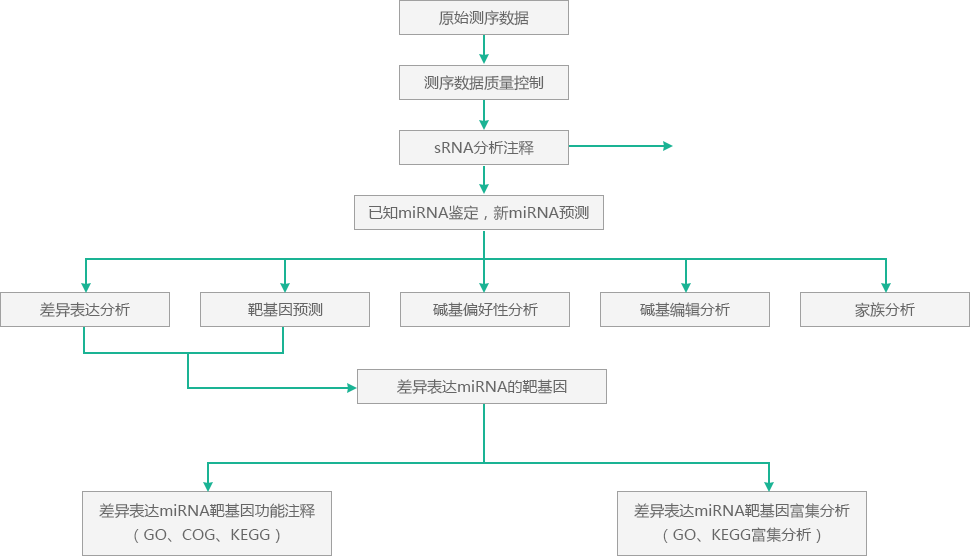
sRNA基本分析以新一代高通量转录组测序(RNA-Seq)数据作为输入,测序得到的原始序列含有接头序列或低质量序列,为了保证信息分析的准确性,需要对原始数据进行质量控制,得到高质量序列(即Clean Reads),原始序列质量控制的标准为:
1.去除低质量Reads(质量值低于30的碱基所占比例超过20%);
2.去除未知碱基N含量大于等于10%的Reads;
3.截除3’端接头和Barcode序列;
4.去除短于18或长于30个核苷酸的序列。
对于得到的Clean Reads,利用Bowtie[1]软件,将其分别与Silva数据库、GtRNAdb数据库、Rfam数据库和Repbase数据库进行序列比对,过滤核糖体RNA(rRNA)、转运RNA(tRNA)、核内小RNA(snRNA)、核仁小RNA(snoRNA)等ncRNA以及重复序列,获得包含miRNA的Unannotated reads。
利用miRDeep2软件[2]将Unannotated reads与参考基因组进行序列比对,获取在参考基因组上的位置信息,即为Mapped Reads。
针对miRNA的生物特征,对于比对到参考基因组的序列,利用miRDeep2软件进行已知及新的miRNA鉴定。miRDeep2软件将18 30nt的核苷酸序列比对到miRBase数据库中特定物种上,鉴定该物种已知的miRNA;过滤获得的未比对到参考基因组,通过碱基数目延伸,进行miRNA结构预测,获得新的miRNA。
miRNA的鉴定和预测完成后,接下来是对各样本中miRNA进行表达量的统计,并用TPM算法[3]对表达量进行归一化处理。TPM归一化处理公式为:

公式中,readcount表示比对到某一miRNA的reads数目;Mapped Reads表示比对到参考基因组上的reads数目。
利用TPM算法对表达量进行归一化处理后,能够消除不同样品间测序量差异的影响,科学、准确的比较各样品间miRNA的差异表达情况。
检测差异表达miRNA时,需要根据实际情况选取合适的差异表达分析软件。DESeq[4]适用于有生物学重复的实验,可以进行样品组间的差异表达分析,获得两个生物学条件之间的差异表达miRNA集;对于没有生物学重复的实验,则使用IDEG6[5]进行差异表达分析,获得两个样品之间的差异表达miRNA。
在差异表达miRNA检测过程中,使用差异倍数(Fold Change,FC)和错误发现率(False Discovery Rate,FDR)作为筛选标准。FC表示两样品(组)间表达量的比值。由于miRNA的差异表达分析是对大量的miRNA表达量进行独立的统计假设检验,会存在假阳性问题,因此在分析过程中,采用了公认的Benjamini Hochberg校正方法对原有假设检验得到的显著性p值(p value)进行校正,并最终采用FDR作为差异表达miRNA筛选的关键指标。
根据已知miRNA和新预测的miRNA与对应物种的基因序列信息,对于植物物种,我们使用TargetFinder软件[6]进行靶基因预测;对于动物物种,用miRanda[7]和RNAhybrid软件[8]进行靶基因预测,对两个软件的预测结果取交集。
靶基因预测完成后,我们会使用BLAST软件将预测到的靶基因与NR[9]、Swiss Prot[10]、GO[11]、COG[12]、KEGG[13]、KOG[14]、Pfam[15]数据库比对,获得靶基因的注释信息。
小RNA在疫霉菌感染大豆中的防御机制解析
本研究对大豆抗病材料 Williams82 和感病材料Harosoy接种大豆疫霉菌,未接种和接种后 8 小时根部进行小 RNA测序,每个样品测序数据量 10-13M。共鉴定到已知miRNA 324 个,属于 109 个miRNA家族。预测到新miRNA 54个,与已有大豆降解组数据比较发现 36 个miRNA具有较高可信度。miR-1507 和 miR-2109 调控 NB-LRR 基因的表达;NB-LRR 和 PPR 基因产生的phasiRNA在侵染后显著上调。这些miRNA和phasiRNA的生物学功能证明在疫霉菌感染大豆中的防御中有重要的调控作用。
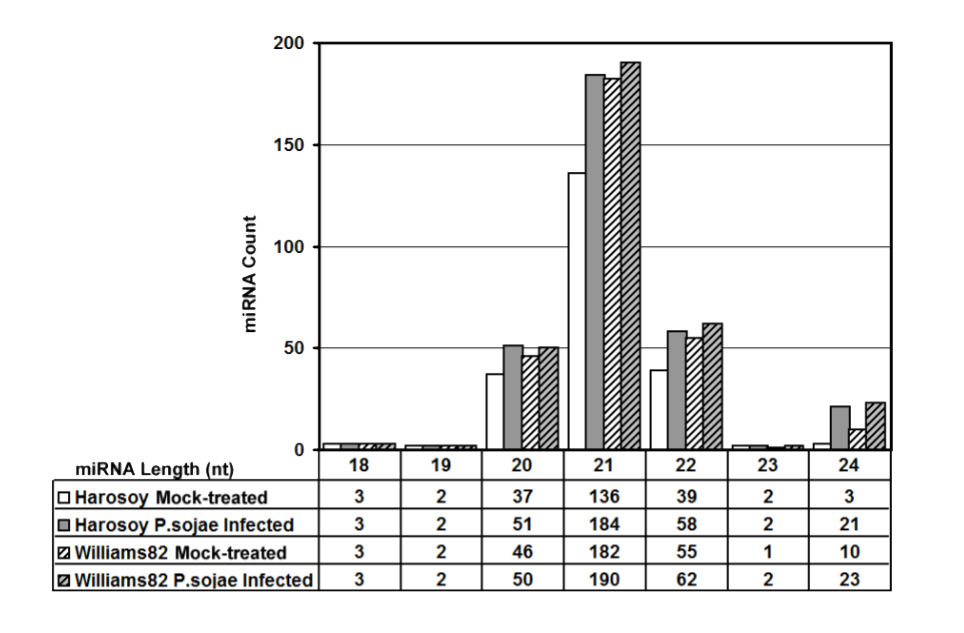
图1.miRNA 长度分布统计
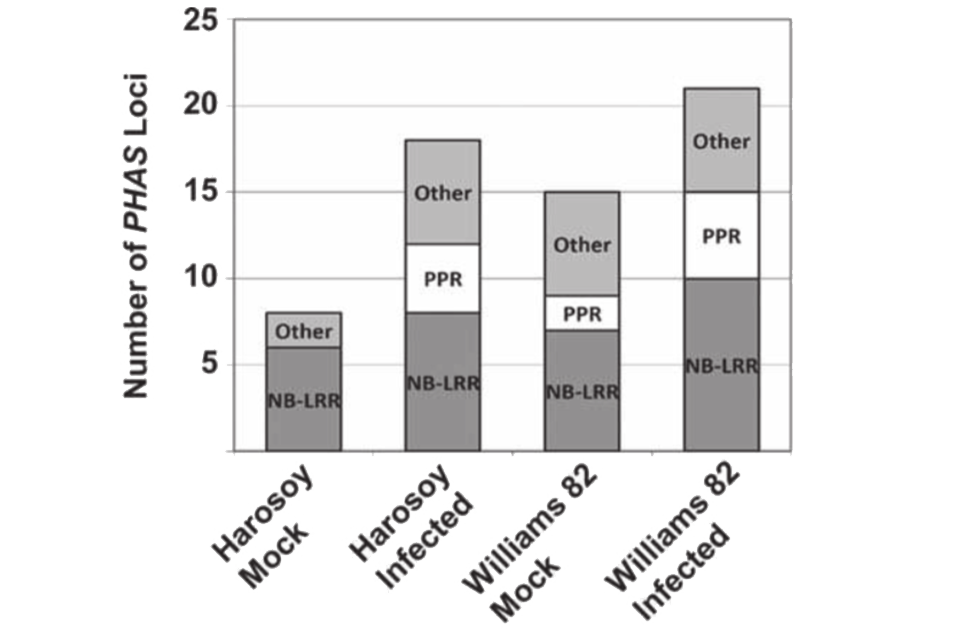
图2.phasiRNA 及其前体基因统计