

基于已知的基因组序列和注释信息,以新一代高通量转录组测序(RNA-Seq)数据作为输入,根据测序数据与参考基因组的序列比对,识别新的转录位点(新基因)、新的可变剪接事件,并对新旧基因进行结构分析、表达定量和差异表达分析。测序数据质量评估;测序数据与所选参考基因组的序列比对;确定外显子/内含子的边界,分析基因可变剪接情况;发掘未注释的基因区和新的转录本;识别转录区的SNP位点;修正已注释的5’和3’端基因边界;定量基因和转录本表达水平,识别不同样品(组)之间显著差异表达的基因并对其进行功能注释和富集分析。 测序数据量为6Gb的6个小鼠样品分析耗时约5天。
转录组是连接基因组遗传信息与生物功能的蛋白质组的纽带,转录水平的调控是最重要也是目前研究最广泛的生物体调控方式。转录组测序能够对样品任意时间点或任意条件下的转录组进行测序,拥有精确到单个核苷酸的分辨率。能够动态反映基因转录水平,同时鉴定和定量稀有转录本和正常转录本,并且提供样品特异的转录本序列结构信息。
目前转录组测序技术已广泛应用于农学、医学等各个研究领域,包括动植物发育调控、环境适应、免疫互作、基因定位、物种遗传进化及肿瘤与遗传病检测等。
有参转录组测序分析平台可以进行标准分析和个性化分析,其中标准分析包括:差异表达基因分析、基因结构分析、新基因分析等。设定参数后点击提交进行分析,分析完成后在流程定制页面下生成标准化结题报告,实现一键式生成。
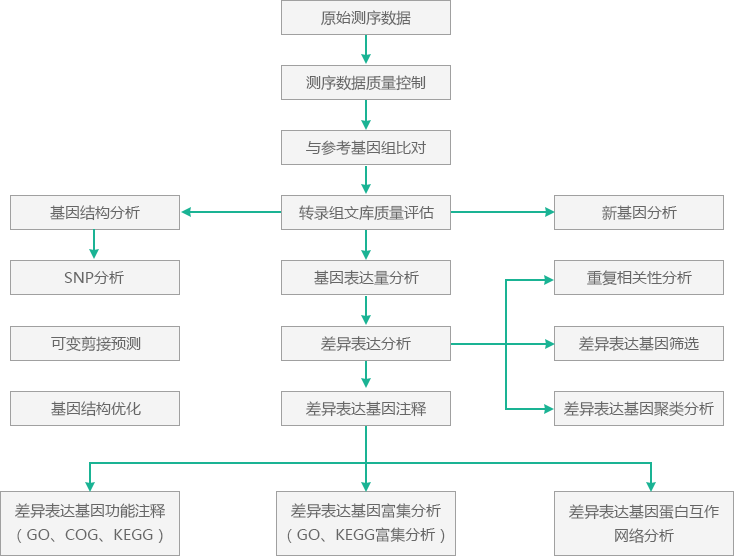
分析平台分析流程以BMK自主研发的程序对测序数据进行质量评估为开始,包括数据量、数据质量Q30等的评估。
做完数据质量评估之后,使用软件TopHat2将测序Reads比对到参考基因组上,TopHat2[1]是专门的RNA-Seq比对软件,它通过分割Reads二次比对参考基因组,不仅提高了测序数据的利用率,还为后续新可变剪接事件的预测提供了基础。
基于各样品Reads与参考序列的序列的TopHat2比对结果,使用软件Cufflinks[2]进行转录本的拼接、表达定量。通过与已知基因组注释文件的比较,识别新的转录组区域,即新基因。通过软件BLAST[3]将新基因与各数据库进行序列比对,获取新基因的注释信息。
基于各样品reads与参考基因组序列的TopHat2比对结果,使用软件SAMtools[4]识别测序样品与参考基因组间的单碱基错配,查找基因区潜在的SNP位点。
根据与基因原有的剪接模型进行比较,使用Cufflinks软件从跨内含子Reads中预测新的可变剪接事件,并用软件SpliceGrapher[5]对其进行可视化。
接着根据基因在不同样品中的表达水平,使用软件DESeq[6]或EBSeq[7]进行差异表达分析,并通过指标FDR和FC筛选差异表达基因。
最后,提取各差异表达基因集的注释信息,使用Fisher精确检验、topGO[8]等进行差异表达基因集的GO节点或KEGG通路富集分析。
案例解析
The identifiation of Cucumissativus Glabrous 1 (CsGL1)required for the formation of trichomes uncovers a novelfunction for the homeodomain-leucine zipper I gene
研究背景
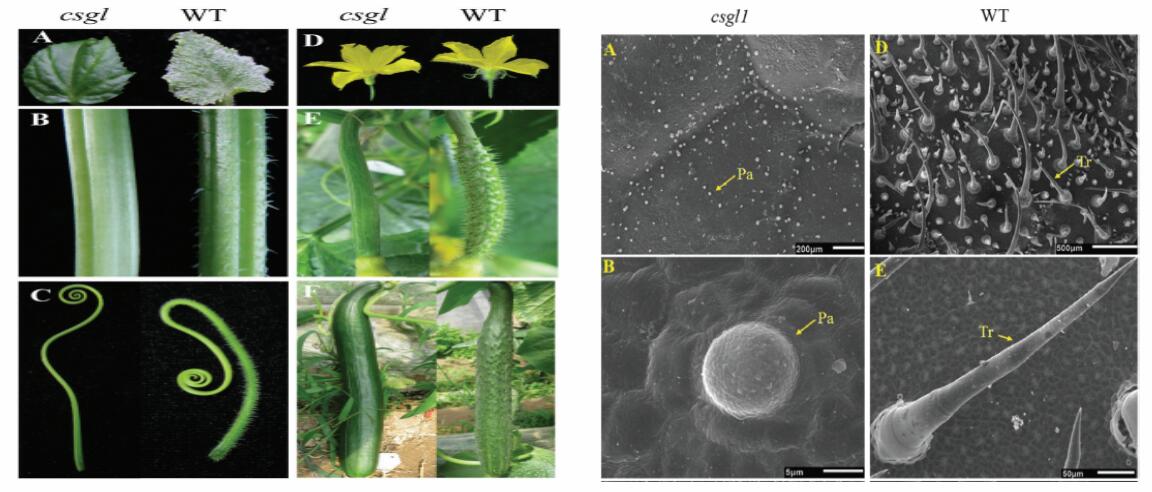
csgl1突变型与野生型两种黄瓜的表型存在很大差异,其中突变型黄瓜的茎、叶、卷须、花萼、子房均无毛,而野生型的都均有毛。本研究想通过转录组测序对两种黄瓜的表型差异进行基因水平的研究。
实验设计
取两种黄瓜品种的叶片,进行RNA提取并进行测序文库构建,最后基于百迈客HiSeq2500平台,进行单端50bp测序,每个样品得到5.7M clean reads。
信息分析
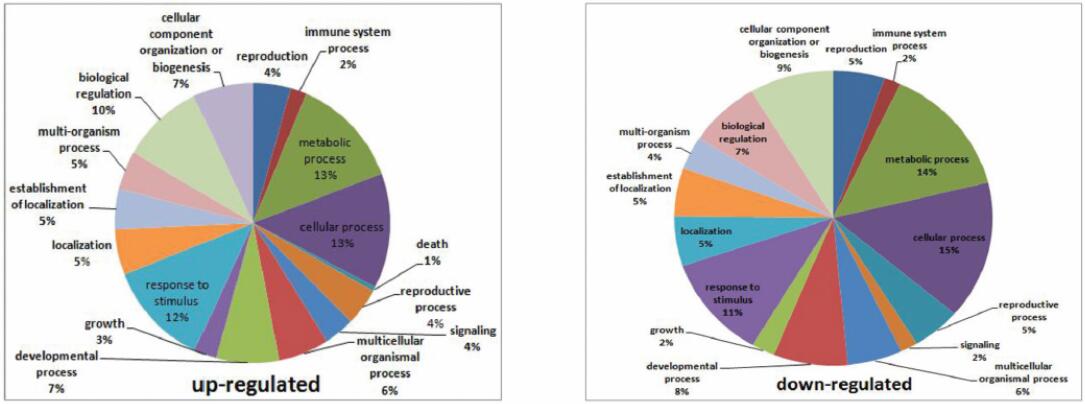
1. 差异表达分析:基于百迈客云平台有参转录组分析平台,共检测到470个差异基因,其中突变体中有269个下调,201个上调,可见CsGL1对黄瓜整个基因表达模式有重要影响。进一步对差异基因进行功能分析,其中有350个注释到细胞过程,335个注释到代谢过程,且与黄瓜叶毛状体、果实瘤刺及花毛状体形成相关。
2. 图位克隆:从F2代选择26个无毛的个体进行定位,将CsGL1基因定位在3号染色体的SSR21054及SSR117两个标记之间;利用2个STS及3个CAPS标记对32重组自交系个体进行精细定位,最终用STS-2 及CAPS-1将CsGL1基因定位在79.7 kb的区间内,与葫芦基因组数据库比较,注释到13个候选基因。
分析结果:分别对突变体与WT个体中13个候选基因进行比较,发现其余12个基因序列无差别,仅Csa3G748220基因在隐性突变体中有2649bp碱基的缺失。表明Csa3G748220是唯一的候选基因。