高通量测序技术的出现,使全世界产出的测序数据出现了爆炸式增长,这些数据存放在或大或小的数据库中,区域性的大数据库包括NCBI、ENA/EBI、DDBJ等,今天我们重点给大家介绍下NCBI的SRA数据库。
Part 1 | SRA数据库介绍
SRA(Sequence Read Archive)是NCBI中专门用于存放原始
高通量测序数据的一个子库,收录了各种二代、三代测序仪产生的数据,与ENA/EBI、DDBJ间共享原始测序数据。

INSDC(International Nucleotide Sequence Database Collaboration)成员间共享测序数据
有过数据上传经历的童鞋应该对SRA并不陌生,上传数据前我们一般要创建BioProject、BioSample,用于详细说明项目信息、样品信息;并通过SRA的Experiment、RUN描述建库测序相关信息,如建库类型、测序仪器、单双端等;下图概括出了几者之间的关系。
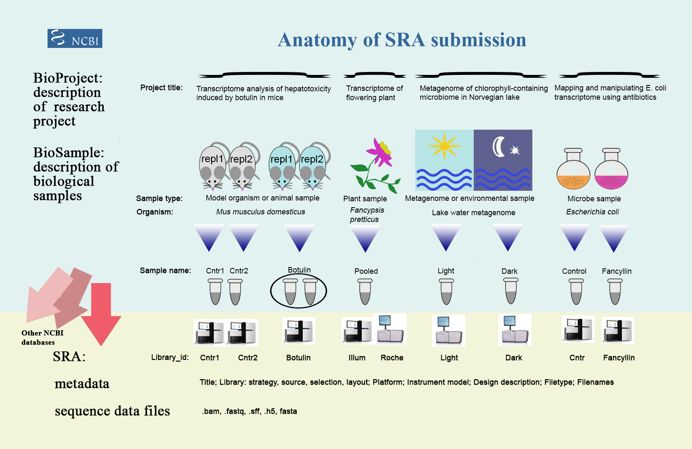
https://www.ncbi.nlm.nih.gov/sra/docs/submitmeta/
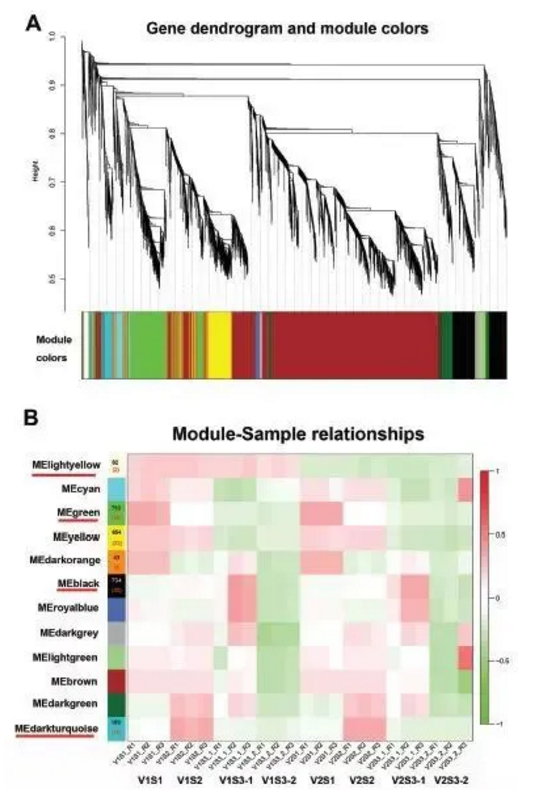
SRA上传和检索数据时,我们会遇到各种各样的编号,这些编号间的对应关系通过下表我们可以理清。项目和样品信息首先会存放在BioProject和BioSample数据库中,得到类似PRJNA和SAMN的编号;在SRA数据库中也会对项目和样品进行编号,分别以SRP和SRS作为前缀,并与BioProject和BioSample中对应;其余SR开头的编号都属于SRA数据库。

SRA数据库中各种编号对应表
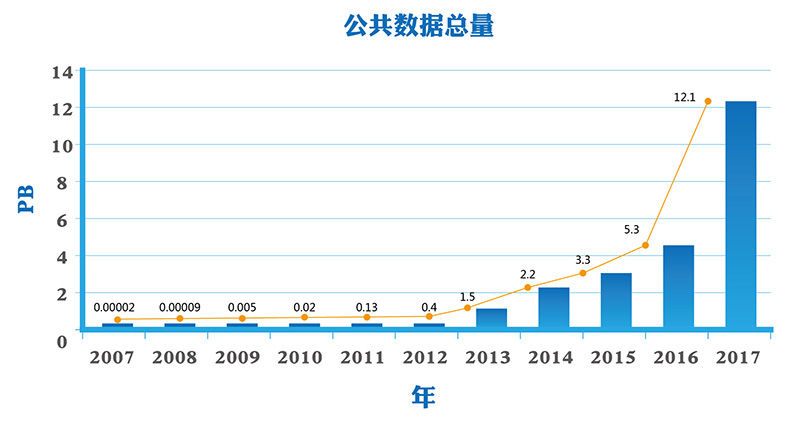
SRA数据库中存储的是高度压缩后的sra格式数据,截止到目前,SRA中已经累计存储了超过
20P碱基数据,而且每年仍在以极快的速度增长。
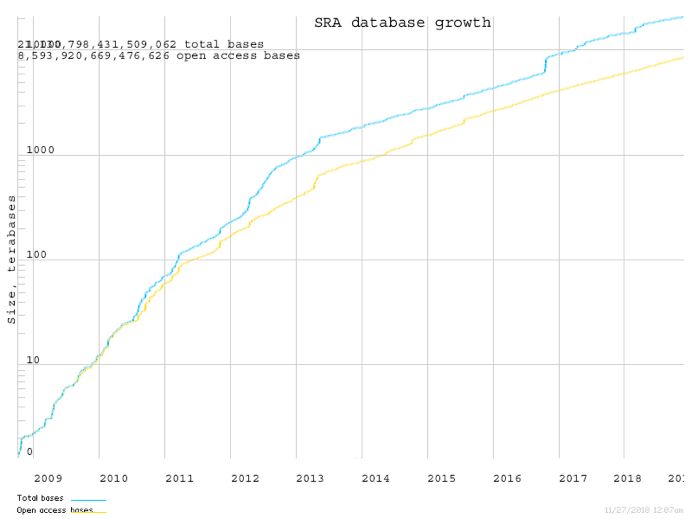
SRA数据量增长图(纵坐标代表sra格式文件大小,单位TB;横坐标代表年;蓝线代表总数据量)
Part 2 | SRA数据库中疾病相关数据统计
在SRA数据库的愿景中,除了进行原始测序数据的保存之外,还有一个目的就是希望这些数据可以被再次利用,得出新的发现。但是目前这些数据就像宇宙中无法被探测的暗物质,无人问津。
https://www.ncbi.nlm.nih.gov/sra/docs/
既然已经有如此多的公共数据,我们应该充分挖掘,不仅可以产出新发现,也可以有效降低科研成本。俗话说的好,知己知彼,百战不殆。要想充分利用这些公共数据,我们首先需要对这些数据有更加深刻的认识,于是我们针对热点研究疾病,统计了不同测序类型的数据量,以及项目数和样品数,想了解其他疾病数据量情况的童鞋可以文末留言,我们统计好之后发送给您。
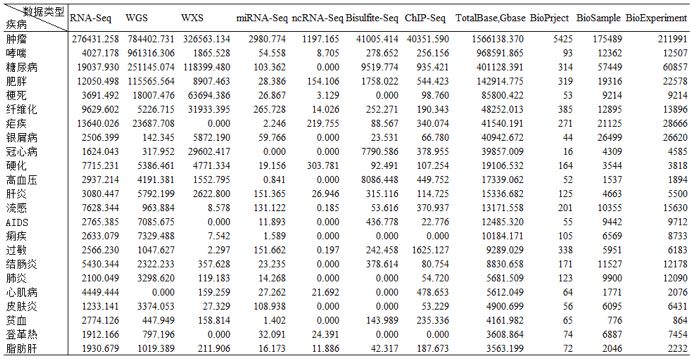
热点研究疾病数据统计(单位:Gbase)
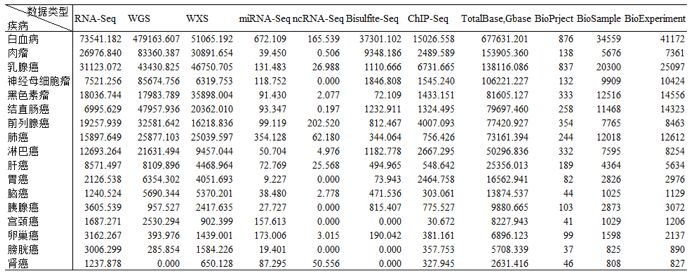
热点癌症数据统计(单位:Gbase)
Part 3 | 公共数据使用策略
如此多的数据,该怎样去利用,我们整理了一些思路,供大家参考。
策略一:数据整合,增大样本量
以研究疾病相关基因表达为例,可以整合多个项目中的RNA-Seq数据(也可以结合自己的数据,增大样本量),计算基因表达量,并筛选疾病组织和正常组织间差异表达的基因;
再针对差异表达基因进行共表达分析,获得共表达基因集;然后进一步对这些基因的功能、所属通路进行分析,从而更完整的描述出疾病发生的机理。
策略二:多种疾病间横向比较
以研究肺癌患者中S100A4基因的差异表达为例,通过下载其他类型癌症如:胸腺癌、恶性间皮瘤的RNA-Seq数据,并分析该基因在这两种癌症中的差异表达情况,如果与肺癌中有相同的差异表达趋势,则可以增强我们结论的说服力。
策略三:不同水平间横向比较
分析不同水平的数据,如:细胞水平、组织水平、动物模型上目标基因的差异表达情况,增强分析结论的说服力。
策略四:不同类型数据间联合分析
我们只自测了mRNA数据,但是想了解miRNA对于mRNA的调控,那我们可以下载对应疾病的miRNA类型的数据,通过两者的联合分析,更深入的了解疾病发生的机理。
公共数据使用看似很困难,需要下载、转换格式、生信分析,目前百迈客云(www.biocloud.net)已经集成了SRA数据检索、下载、转换和分析,我们录制了一个短视频,展示了如何通过简单的鼠标点击高效完成以上所有工作,详情:
http://live.biocloud.net/open/course/10