
08月01 转录调控研究中公共数据整合分析思路
分析思路1
共表达分析中,整合大量相关公共样本测序数据,可构建出相较于常规样本量下可靠度更高的基因共表达网络,从而基于该网络进行更加准确的后续分析:a)预测目标转录因子的下游调控基因、目标调控网络中的关键转录因子;b)预测ncRNA与mRNA之间的靶向关系;c)基于网络中已知功能基因推测同网络中其他功能未知基因功能;e) 将每个共表达模块分别作为一个整体,计算其与各组织或各发育时间点之间的相关性,建立各组织相关或各时期相关基因表达网络……
思路解析:
一般认为,在功能上具有相关性的基因在生物体内具有相似的表达模式,比如转录因子与其下游调控基因、lncRNA与其反式调控的靶基因、处于同一代谢通路的基因等在功能上都具有相关性,因此,研究者可根据每个基因的表达模式,来判断上述几种相互关系。
生信分析中,研究者可使用共表达分析的方法,基于转录组测序数据,分析每个基因的表达模式,最终将不同的基因划入各自所在的表达模式网络中,常用的共表达分析方法主要有两种WGCNA与k-means,大样本量下(15组以上的样本)建议使用WGCNA,该算法相较于K-means,采用对相关系数取幂加权处理、考虑两个基因间的间接相关等优化算法,使得构建的共表达网络更加符合基因调控网络的无尺度网络分布(scale-free networks)、基因间可间接调控等特征。
无论是使用哪种方法进行共表达分析,从统计学的角度来看,样本量越大,预测的基因表达模式分辨率更高,构建的共表达网络也就越可靠。受限于项目经费,很多研究者便借助公共数据库扩充这类共表达分析中的样本量。
支持文献思路概述:
a) Jennnifer等人鉴定了Specialized Metabiolic(后面简称SM)代谢通路相关基因,此类基因种类较少,且序列保守性较低,难以通过常规的序列同源比对的方法预测,因此研究者采用了基于基因表达量的共表达分析方法,参考少数已知功能基因,鉴定存在于不同植物中的SM代谢相关基因以及SM代谢网络。研究者搜集了8个植物物种的10个基因共表达数据集合的21,876个实验的基因芯片和RNA-seq公共数据,构建了各个物种中高可靠度的基因共表达模块。为了说明本研究鉴定SM途径方法的可靠性,筛选了甲硫氨酸来源的脂肪族硫代葡萄糖苷生物合成途径(metGSL)及基因,与鉴定的共表达基因模块进行比较分析。在拟南芥中,共表达基因模块鉴定了metGSL生物合成每一步的基因,以及一个特异的转运子和3个转录因子。比如,在最小的N1(共17个基因)网络中,metGSL途径的34个酶基因中14个均在这个模块中,该模块中只有3个基因是功能上未鉴定属于metGCL的。在网络中,还发现参与metGSL相关生化过程的一些基因,如激酶APK1和APK2、细胞色素P450基因CYP79B2和CYP79B3。因此,利用该研究中建立的共表达网络可较为准确预测SM代谢通路相关基因,该成果发表于The Plant Cell杂志【文献详细解读见附件1】。
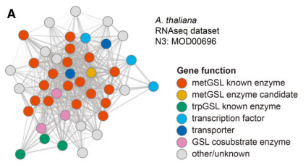
共表达基因模块重现拟南芥metGSL生物合成途
b)Yu C等人,为了揭示与玉米子叶发育各个时期相关的转录因子-调控基因(之后简称TF-TFBS)调控网络,利用公共数据,将样本量从9组个扩充至22组,进行WGCNA分析,建立了与玉米子叶发育各个时期相关共表达模块。基于共表达信息,并参考基因GO注释、TF-TFBS数据库(TRANSFAC、JASPAR、AthaMap等),总共得到176个TF-TFBS,成果发表于PNAS杂志。【原文题目见附件2】
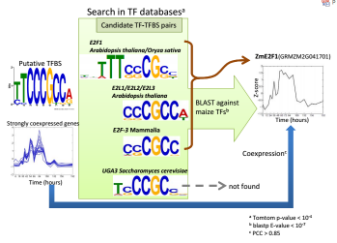
TF-TFBS预测过程
c)Wen Z等人,为了鉴定与大鼠各个发育时期各个组织相关的ncRNA及其与mRNA共同参与的调控网络,从GEO数据库中下载得到Ying等人上传的原始转录组测序数据(GSE53960),数据集中包含来自四个发育时间点的32只大鼠的320个bodymap样本的转录组测序数据。首先基于测序数据进行转录本组装、定量、ncRNA鉴定、差异表达等前期分析,然后基于基因定量结果,使用方差分析(analysis variance,ANVOA)鉴定不同的发育时期、不同的组织部位中或性别间显著差异的基因(Benjamin-Hochberg corrected p-value < 0.05),鉴定获得的差异基因即为时期相关、组织相关或性别发育相关基因,之后利用WGCNA分别对上述各个基因集构建共表达网路模块,最终鉴定得到32个器官发育相关模块、4个性别发育相关模块、14个发育时期相关模块。该成果发表于Scientific Reports杂志【文献详细解读见附件1】。
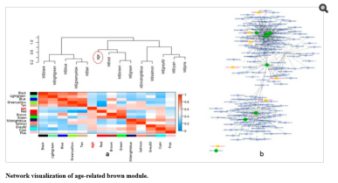
发育时期相关共表达模块
d)LiJ等人为了全面鉴定猪的长链非编码RNA(lincRNAs)和探索lincRNAs在猪植入前胚胎发育(PED)过程中可能发挥的作用,从NCBI-SRA数据库中下载得到五个猪RNA-Seq数据集。基于测序数据进行转录本组装和筛选后获得了7,618个lincRNAs。在分析了猪lincRNAs的结构特点、表达模式、组织特异性和顺式作用后,对胚胎发育阶段相关lincRNAs和mRNAs进行了WGCNA分析,鉴定出了23个共表达模块,其中5个显示发育阶段特异性。qRT-PCR分析4细胞阶段特异性模块中的枢纽基因集后发现了两个与PED密切相关的lincRNA:TCONS_00166370 和TCONS_00020255。该成果发表于Scientific Reports杂志【文献详细解读见附件1】。
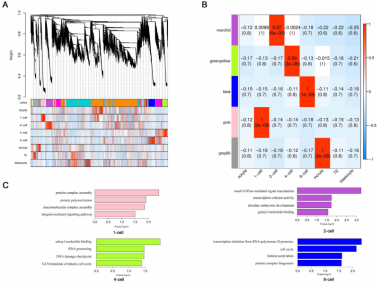
共表达网络分析PED相关lincRNAs
分析思路2
研究某一类基因的转录水平在不同处理下(或不同组织部位间、或不同发育时间点等)的变化规律,可整合多个类似研究中的公共测序数据来共同揭示该变化规律,使结果更加可靠。
思路解析:
该思路相对比较简单,但是若论点新颖,多个项目或多个物种的数据均对此论点支持,该分析也可单独成文;该分析也可作为对常规转录调控分析的补充,从常规分析得到的一些相关基因中挑选一些关键基因,在其他物种、其他类似项目中寻找对该类关键基因在该项目中某种变化规律的支持证据,可以提升常规分析的广度。
支持文献思路概述:
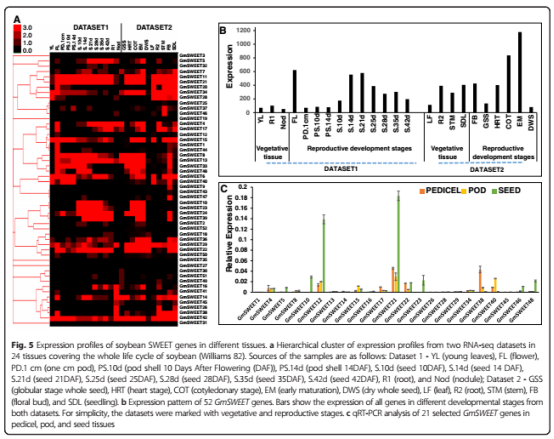
a)sweet基因家族编码一类外排转运蛋白(efflux transporter),与糖运输、韧皮部装载、宿主-病原菌互作、生殖组织发育相关,这类基因的研究主要集中在水稻与拟南芥中,而在大豆中的研究几乎是空白。
Gunvant P等人搜集了公共数据库中两个大豆RNA-seq数据集,分别包含14、10个样本,均涵盖生殖组织(花、花芽、种子等)与营养组织(根、茎、幼苗等)。研究者首先基于水稻、拟南芥的sweet家族基因序列,通过blast比对,在大豆的基因组中鉴定得到52个大豆sweet家族基因,并对该类基因在染色体上的分布和编码蛋白的domain结构与其他13个物种(涵盖单子叶、双子叶、藻类、苔藓类)中sweet基因家族的进化关系进行了简单分析。
两个数据集中大豆的RNA-seq数据分析结果表明,大部分大豆sweet家族基因在种子和花中转录上调,所有sweet基因在到达种子灌浆期前转录水平一直上调,之后到达种子成熟前一直下调,这与之前的在其他物种中研究得到的该基因家族与生殖组织发育相关的结论是一致的。该成果发表与BMC Genomics杂志。
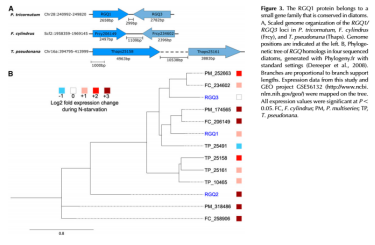
b)Matthijs M等人通过分析自测RNA-seq数据在三角褐指藻发现了一类可响应氮胁迫的新型转录因子RGQ1,为了进一步验证该转录因子是否在其他硅藻中存在并且也同样具有响应氮胁迫的功能,研究者从公共数据库中下载到了其他两种硅藻类似研究中的数据,发现,RGQ1在这两种硅藻中也存在,且参与了对氮胁迫的响应。成果表于Plant Physiology杂志。【详细解读结果见附件1】
其他分析思路
整合同一物种公共测序数据,构建物种完备转录本序列参考集,用于后续功能分析。
支撑文献:
- Iyer MK et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015
文献概要:整合来源25个项目,18个组织,7256个样品RNA测序数据,使用cufflinks分别重构各组织转录本,之后使用用户自己开发的meta-assembly算法找出各组织中高丰度转录本,最后使用cuffmerge对各样本组装得到的转录本进行合并,最终建立高质量的人类转录本序列参考集,用于后续功能分析。
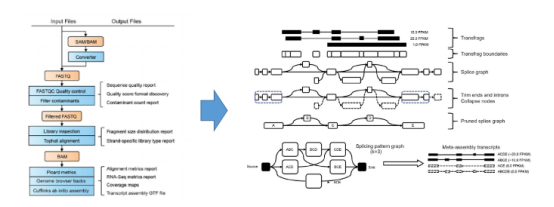
- Wang M et al. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015
文献概要:整合170多个棉属RNA-seq数据集,以海岛考基因组为参考,使用tophat+cufflinks流程进行转录本重构(各样本分开组装)并预测其中的lncRNA,之后进行棉纤维发育相关lncRNA鉴定。
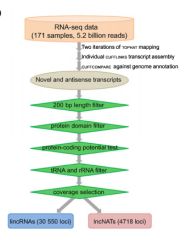
mRNA-ncRNA联合分析中,利用公共数据补充其中一种类型的RNA测序数据
支撑文献:
- Liu X et al. MicroRNA-mRNA regulatory networking fine-tunes the porcine muscle fiber type, muscular mitochondrial respiratory and metabolic enzyme activities. BMC Genomics. 2016
文献概要:联合之前项目的高肉品与低肉品猪mRNA测序数据与本次项目中的miRNA测序数据,并通过pearson相关系数建立miRNA与猪肉品质各项指标之间的联系,最终建立与猪肉品质各项指标相关的mRNA-miRNA调控网络。
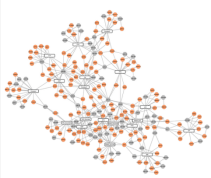
- Yin H et al. Phylogenetic tree-informed microRNAome analysis uncovers conserved and lineage-specific miRNAs in Camellia during floral organ development.J Exp Bot.2016
文献概要:研究者取红杜鹃山茶花的叶、雄蕊、雌蕊心皮、花瓣、花芽5个部位进行了miRNA测序,之前项目中组装得到的红杜鹃山茶花的转录本序列为参考,进行了novel miRNA的预测。之后通过miRNA表达模式的分析,鉴定到了两类分别倾向在雄蕊或雌蕊中特异高表达的miRNA,进一步利用miRNA靶基因的功能信息,揭示了这些miRNA在花器官发育过程中发挥的生物学功能。
附件2公共数据整合分析文献列表
整合公共数据建立物种基因共表达网络
[1] Wisecaver JH et al. A Global Coexpression Network Approach for Connecting Genes to Specialized Metabolic Pathways in Plants. Plant Cell. 2017
[2]Wen Z et al.Expression profiling and functional annotation of noncoding genes across 11 distinct organs in rat development.Sci Rep. 2016
[3] Li J et al. Identification and functional analysis of long intergenic noncoding RNA genes in porcine pre-implantation embryonic development. Sci Rep. 2016
[4] Yu C et al. Transcriptome dynamics of developing maize leaves and genomewide prediction of cis elements and their cognate transcription factors. Proc Natl Acad Sci . 2015
[5] Khan FA et al. Analysis of Bos taurus and Sus scrofa X and Y chromosome transcriptome highlights reproductive driver genes. Oncotarget. 2017
利用公共数据,研究基因家族转录水平变化规律
[6] Patil G. et al. Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics. 2015
[7]Matthijs M et al.Profiling of the Early Nitrogen Stress Response in the Diatom Phaeodactylum tricornutum Reveals a Novel Family of RING-Domain Transcription Factors. Plant Physiol. 2016
基于公共数据建立物种完备转录组本参考序列
[8]Wang M et al. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015
[9]Iyer MK et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015
[10]Hong Y, et al. Genome-wide identification and characterization of long intergenic noncoding RNAs and their potential association with larval development in the Pacific oyster. Sci Rep, 2016.
[11] Li J, et al. Identification and functional analysis of long intergenic noncoding RNA genes in porcine pre-implantation embryonic development. Sci Rep,2016.
基于公共数据进行ncRNA-mRNA联合分析
[12] Xu W et al. Genomic DNA Methylation Analyses Reveal the Distinct Profiles in Castor Bean Seeds with Persistent Endosperms. Plant Physiol. 2016
[13] Liu X et al. MicroRNA-mRNA regulatory networking fine-tunes the porcine muscle fiber type, muscular mitochondrial respiratory and metabolic enzyme activities. BMC Genomics. 2016
其他分析思路
[14] Sudmant PH, et al. Meta-analysis of RNA-seq expression data across species, tissues and studies. Genome Biol, 2015.
[15] Lu L, et al. The goose genome sequence leads to insights into the evolution of waterfowl and susceptibility to fatty liver. Genome Biol, 2015.
[16] Shin SC, et al. Dynamic shifts in occupancy by TAL1 are guided by GATA factors and drive large-scale reprogramming of gene expression during hematopoiesis. Genome Res, 2014
[17] Xie D, et al. Rewirable gene regulatory networks in the preimplantation embryonic development of three species. Genome Res, 2010