
01月13 微生物分析平台个性化分析更新
功能预测分析
基于样品中的物种组成及丰度信息推测样品中表型类型和功能组成及差异。包含PICRUSt2功能预测、FAPROTAX功能预测、BugBase表型预测、Tax4Fun2功能预测和真菌FunGuild表型预测。
一. PICRUSt2功能预测
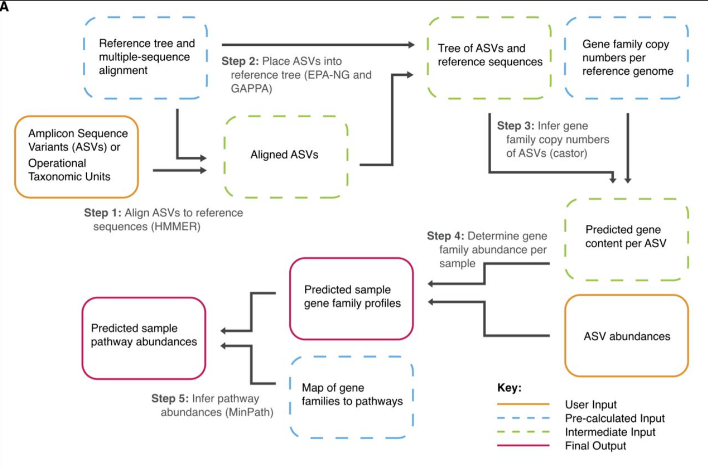
PICRUSt1软件依赖于Greengene数据库进行物种比对、功能数据输出。而Greengene数据库在2013年之后就停止了更新,距今为止已有7年。随着时间的推移,大量微生物基因组数据测序获得,而停止更新的Greengene数据库限制了PICRUSt的功能预测范围。对于近年来测序获得微生物功能功能信息无法进行预测,满足不了当前的研究需求,为了补上这块满足科研工作者的需求,PICRUSt团队于近期升级了软件,正式公布了PICRUSt2。
1)将待预测的OTU代表序列置于软件中已有的系统发育树中,而不是直接对OTU序列进行分类学注释;
2)不再基于GreenGene 16S数据库进行功能预测,其用于预测的参考基因组数据库相比先前也已扩大了10倍以上
参考文献:Douglas G M, Maffei V J, Zaneveld J, et al. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv, 2019.
功能组成分析:统计各样品在不同分类层级上的功能组成。
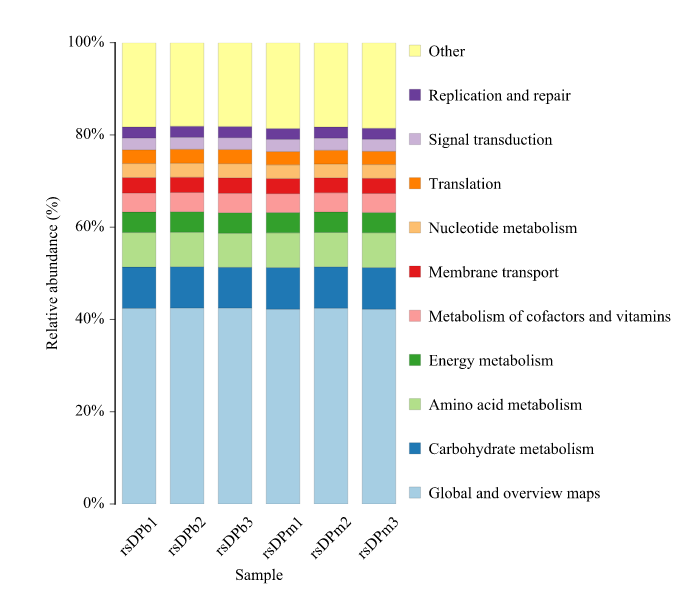
注:横坐标为样品名称;纵坐标为功能相对丰度百分比。
功能差异分析:统计各样品或者各组在不同分类层级上的功能差异。
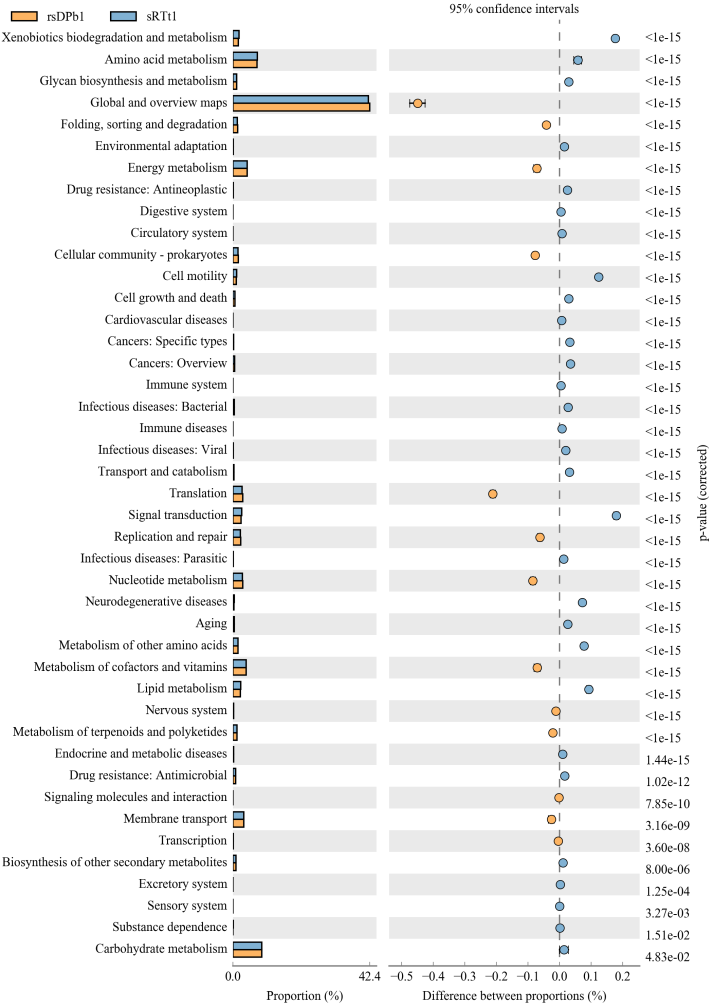
注:图中不同颜色代表不同的样品或分组。左图所示为不同功能在两个样品或者两组样品中的丰度比例,中间所示为95%置信度区间内功能丰度的差异比例,最右边的值为校正后p值。
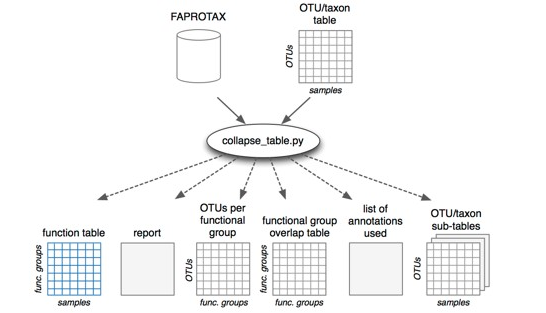
二. FAPROTAX功能预测
FAPROTAX较适用于对环境样本的生物地球化学循环过程(特别是碳、氢、氮、磷、硫等元素循环)进行功能注释预测。FAPROTAX是根据已发表的文献手动构建的数据库,它把原核微生物的分类和代谢等功能对应起来,目前收集自4600多个原核微生物的80多个功能分组7600多条功能注释信息。
参考文献:Louca S, Parfrey L W, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome[J]. Science, 2016, 353(6305): 1272-1277.
三. BugBase表型预测
BugBase是一种预测复杂微生物组内功能途径的生物水平覆盖以及生物可解释表型的方法。BugBase首先通过预测的16S拷贝数对OTU进行归一化,然后使用提供的预先计算的文件预测微生物表型。包括以下七方面:革兰氏阳性 (Gram Positive)、革兰氏阴性 (Gram Negative)、生物膜形成 (Biofilm Forming)、致病潜力 (Pathogenic Potential)、移动元件含量 (Mobile Element Containing)、氧的利用 (Oxygen Utilizing)、氧化胁迫耐受 (Oxidative Stress Tolerant)。
参考文献:Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, SidiropoulosD,Spear J, Caporaso G, Blekhman R, Knight R, Fink R, Knights D. 2017.BugBase predicts organism level microbiome phenotypes. bioRxiv.
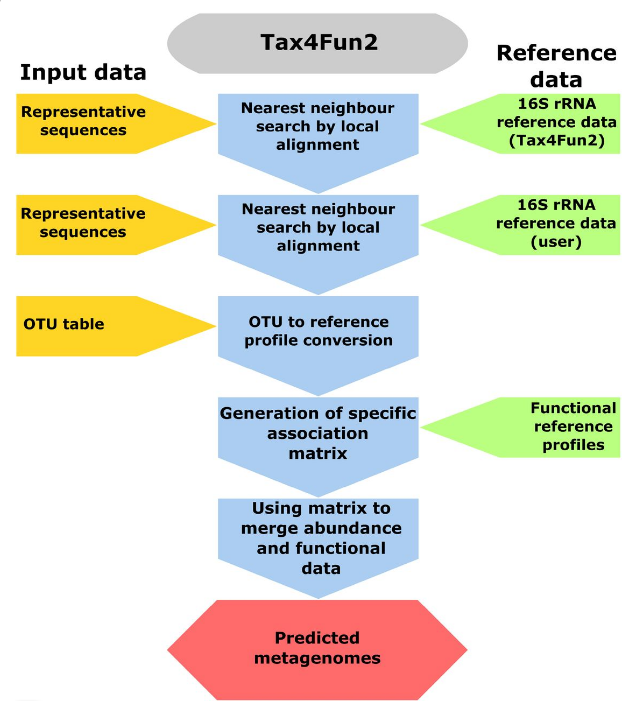
四. Tax4Fun2功能预测
Tax4Fun全面升级为Tax4Fun2.评估微生物群落的功能和冗余度是环境微生物学的主要挑战。Tax4Fun2可基于16S rRNA基因序列快速预测原核生物的功能谱和功能冗余。通过合并用户定义的、特定于栖息地的基因组信息,可以显著提高预测功能图谱的准确性。
优点:
(1)不再局限于仅SILVA的特定版本注释的OTU丰度表,允许直接以OTU代表序列作为输入,通过与指定参考数据库的比对实现物种注释。除了Tax4Fun2提供的已构建好的参考集(相比之前大幅扩大),也允许我们提供自定义的参考集,使用非常灵活。
(2)侧重于原核数据,但也可以合并真核数据。
(3)提供了计算特定功能冗余的方法,对于预测特定功能在环境扰动期间丢失的可能性至关重要。
(4)精度和稳定性显著提升。
参考文献:Wemheuer F, Taylor J A, Daniel R, et al. Tax4Fun2: a R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. bioRxiv, 2018.
五. FunGuild表型预测
FUNGuild(Fungi Functional Guild)是一种可用于由生态协会分类学解析真菌的工具,用简单而一致的方法将大型序列库分类为具有生态意义的类别。根据营养方式将真菌分为12类,然后构建了一个真菌分类和功能分组(guild)之间的数据库,通过这个数据库你就可以对真菌进行功能分类。
病理营养型(pathotroph):通过损害宿主细胞而获取营养(包括吞噬型真菌phagotrophs)。
共生营养型(symbiotroph):通过与宿主细胞交换资源来获取营养。
腐生营养型(saprotroph):通过降解死亡的宿主细胞来获取营养。包括动物病原菌(animal pathogens)、丛枝菌根真菌(arbuscular mycorrhizal fungi)、外生菌根真菌(ectomycorrhizal fungi)、杜鹃花类菌根真菌(ericoid mycorrhizal fungi)、叶内生真菌(foliar endophytes)、地衣寄生真菌(lichenicolous fungi)、地衣共生真菌(lichenized fungi)、菌寄生真菌(mycoparasites)、植物病原菌(plantpathogens)、未定义根内生真菌(undefined root endophytes)、未定义腐生真菌(undefined saprotrophs)和木质腐生真菌(wood saprotrophs)。
参考文献:Nguyen NH, Song Z, Bates ST, Branco, S, Tedersoo L, Menke J, Schilling JS, Kennedy PG. 2016. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology 20:241-248.
MicroPITA分析
在做完16S、18S或ITS等微生物多样性研究后,我们常常还会想进一步了解微生物群落的功能。通常情况下,会采用宏基因组、宏转录组或宏代谢组等方法深入分析,但相对于扩增子测序,宏基因组等测序手段的价格还是相对较高,因此需要从已测完的样本中再挑选合适的样本进行宏基因组测序。可利用microPITA进行样品预测,挑选出合适的样品。该分析是基于大量微生物多样性的数据,根据不同指标筛选出代表性样本,以便于开展开展后续研究。
| 类型 | 方法 | 含义 | 样本特点 |
| 无监督方法 | diverse | 选择α多样性最高的样本 | 生态多样性高 |
| features | 根据目标物种挑选样本 | 针对特定物种 | |
| extreme | 选择β多样性距离最远的样本 | 极端样本 | |
| representative | 最能反映整体距离差异的样本 | 核心样本 | |
| 有监督方法 | Distinct | 根据表型/分组特征,挑选组间β多样性距离最大的样本 | 依据表型/分组特征,选择极端样本 |
| Discriminant | 根据表型/分组特征,挑选离分组中心最近的样本 | 依据表型/分组特征,挑选核心样本 |
参考文献:Tickle TL, Segata N, Waldron L, Weingart U, Huttenhower C. Two-stage microbial community experimental design. ISME J. 2013 Dec;7(12):2330-9. doi: 10.1038/ismej.2013.139. Epub 2013 Aug 15. PMID: 23949665; PMCID: PMC3834858.
群落结构分析
微生物群落生态学的一个主要目标是了解构成跨时空物种丰度模式的过程。确定性和随机性两种类型的过程会影响群落的聚集。确定性过程与生态选择相关,随机过程包括不可预测的扰动、概率性的散布和随机的出生-死亡事件等,这些变化不是由环境决定的适应性结果。通过零模型量化群落的绝对系统发育距离与随机系统发育距离的偏离度,偏离程度越大,群落受确定性因素的影响越大,偏离度越小,群落受随机性因素的影响越大。通常使用βNTI(最近种间亲缘关系指数)以评估不同时空尺度下随机性和确定性过程对微生物群落组装的影响。
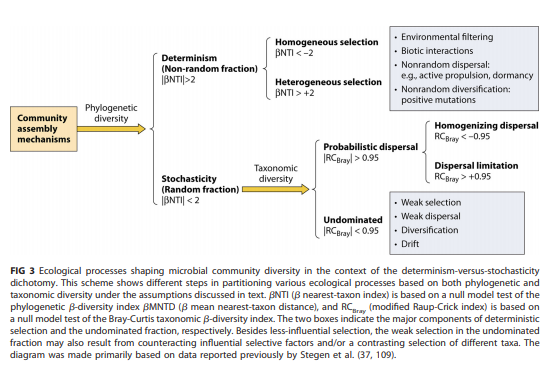
其中,| βNTI |>2表示观察到的两个群落之间的更替主要由选择控制,其中βNTI>+2与变量选择一致,而βNTI<-2表示同质选择。因此,| βNTI |<2意味着一组群落的更替受扩散限制、均匀化扩散或未消除过程的控制。为了理清这些过程,Raup-Crick矩阵(RCbray)基于群落的标准Bray-Curtis矩阵构建,提供有关所观察到的流动程度是否明显偏离预期的信息。这个值等于观测到的Bray-Curtis和零分布之间的偏差,范围是-1到+1。| RCbray |<0.95可以解释为终止过程的影响。反过来,扩散限制加上漂移导致大于预期的周转率(RCbray>+0.95),而RCbray<-0.95则表明群落组成的周转率主要受均匀扩散控制。
参考文献:Jizhong, Zhou, Daliang, et al. Stochastic Community Assembly: Does It Matter in Microbial Ecology[J]. Microbiology & Molecular Biology Reviews, 2017.