

09月01 【成功案例】小RNA和降解组联合测序揭示microRNA调节茶叶(Camellia sinensis)中儿茶素生物合成
小RNA和降解组联合测序揭示microRNA调节茶叶(Camellia sinensis)中儿茶素生物合成
Combined small RNA and degradome sequencing reveals complex microRNA regulation of catechin biosynthesis in tea (Camellia sinensis)
杂志:PLOS ONE
影响因子:2.806
PMID: 28225779
研究背景
MicroRNAs是一种内源性非编码小RNA,在动植物转录后调控及其他生物学过程中有至关重要的作用。基于碱基互补配对机制,miRNAs主要通过直接切割目标mRNA和抑制目标基因转录这两种方式调节目标基因的表达。前期研究表明,miRNA参与多种植物的生长和发育过程,如:根、叶、花的形态发生,信号转导,激素反应和营养代谢等。植物miRNAs主要通过克隆、生物信息预测、高通量测序和Northern 杂交的方法来检测。最常用的技术是高通量测序,不仅经济实惠,而且在低丰度的情况下可以更简单、快速获得大量miRNAs。植物中miRNA主要是通过切割目标基因或者抑制目标基因的表达进行调控,所以目标基因的确定对miRNA功能的分析至关重要。miRNAs目标基因的预测可以通过生物信息的方法,目标基因的鉴定主要通过降解组测序和5’-RLM-RACE方法(RNA连接酶介导的5’cDNA末端的快速扩增),这两种方式还能鉴定miRNA目标基因切割位点,有助于miRNA功能分析。
茶树(Camellia sinensis),富含儿茶素,是风靡全球的非酒精饮料之一。儿茶素是茶叶中重要的组成部分,包括脂型儿茶素(如:ECG和EGCG)和非脂型儿茶素(如:EC和EGC)。EGCG是茶叶中含量最丰富的儿茶素并且具有较强的抗癌效果,可以制约癌细胞的生长,抑制雄激素受体的功能并调控癌细胞的生存,血管的生成和移动。基于他们的分子构成,儿茶素可以分为简单儿茶素和脂型儿茶素。在儿茶素的合成过程中有许多酶的参与,如查耳酮合酶(CHS),查尔酮异构酶(CHI),黄烷酮醇4-还原酶(DFR),花青素合酶(ANS),花青素还原酶(ANR)和类黄酮3,5羟基化酶(F3’5’H)。MiRNA被鉴定为转录和转录后水平基因表达的重要调节因子,并且裂解目标mRNA是植物中基因表达的主要调控方式。
在茶树中,虽然通过高通量测序和芯片技术发现了许多保守的以及新的miRNAs,但是miRNAs直接调控儿茶素生物合成还没有明确的定义。此项研究中,研究人员通过高通量测序鉴定茶叶中保守的以及新的miRNAs,再通过5’-RLM-RACE方法分析潜在的目标miRNAs。通过表达模式分析进一步筛选儿茶素积累过程中潜在的miRNAs。结合5’-RLM-RACE实验和表达模式分析,研究人员发现了一些新的调节儿茶素生物合成途径中基因切割的调控因子。
材料和方法
实验材料
茶树1005,取叶片(包括芽,第一叶,第二叶,第三叶,第四叶,第五叶,成熟叶),进行混合,提取总RNA。
茶树1005,取叶片(包括第一叶,第三叶,成熟叶),进行qRT-PCR表达分析和儿茶素含量测定。
测序平台:Illumina HiSeq 2500 (Biomarker Technology Co.)
技术路线
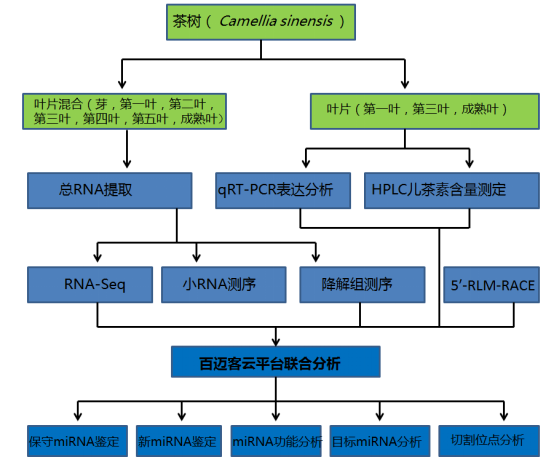
实验结果
1.茶叶中miRNA的初步分析
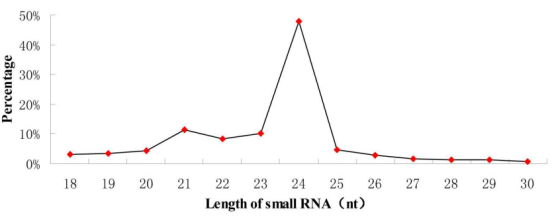
从提取的总RNA中构建smallRNA文库,测序质控后得到19,567,691条clean reads,与数据库比对发现有18.83%的rRNA,1.23%的tRNA,0.01%的small nuclear RNA,0.07%的重复序列,其中79.85%的序列没有功能注释,可以将这些没有功能注释的序列用来做下一步的miRNA的预测和鉴定。根据smallRNA的测序长度分布图可以得出,miRNA的长的主要分布在20-25nt之间,丰度最高的是24nt(47.89%)。这个长度分布和其他栽培茶以及其他植物报道的结果非常相似。
2.茶叶中保守miRNAs的鉴定
将没有功能注释的clean reads与miRBase(植物已知保守miRNA数据库)进行比对,共获得69个保守的miRNAs,分别属于62个家族。根据长度分布发现,这些保守的miRNAs的长度主要集中在18-25nt之间,长度主要集中在24nt(46.385),其次是21nt。
对高通量测序的miRNA片段进行分析,鉴定的保守miRNA中,read values低于1000的占91.3%的比例,其中read values低于10的占56.52%的比例。研究人员还发现了一些高表达(read values大于1000)的miRNA家族,如miR165a和miR396a。另外,在不同家族中的miRNAs表达量差异较大,并且在同一家族中各miRNAs之间的表达量也有较大差异。这些数据表明,不同家族miRNA的表达模式不同,意味着不同的miRNAs在茶树生长发育过程中的作用不一样。
3.茶叶中新miRNAs的鉴定
通过miRDeep2和Mfold软件分析,共鉴定出47个新的miRNAs,进一步对这些新miRNA进行片段长度和表达量分析。新miRNA跟保守miRNA长度分布相似,主要集中在18-25nt之间,最集中的长度是24nt和21nt。另外,新miRNA的表达量比保守miRNA的低,新miRNA中表达量最高的只有4517个read values。
研究人员将这些新的miRNA与其他植物中已知的miRNA进行比对,揭示了许多同系物。除了三个miRNA归为同一个家族外,绝大多数的miRNA可以被分为独立的家族。研究者还对新miRNA的前体进行了分析,发现他们都有一个发卡环结构,长度在84-114bp。
4.GO功能富集和KEGG通路分析目标miRNA
为了研究茶树miRNA的功能,研究人员运用TargetFinder软件和转录序列对上述miRNA进行目标基因预测。结果显示,其中97个miRNA对644个目标基因有调控作用。再将644个目标基因进行GO和KEGG通路富集分析:GO富集分析发现这些目标基因主要富集在细胞、细胞器、细胞膜、连接活性、转运活性、新陈代谢和细胞进程等功能条目;KEGG通路富集发现366个目标基因参与36个代谢网络相关,主要参与糖酵解、柠檬酸循环、氨基酸代谢、光合作用、脂肪酸代谢、嘌呤嘧啶代谢、氧化磷酸化、植物病原体相互作用、初级代谢产物和次级代谢产物过程。
- miRNA靶向基因降解组测序和功能分析
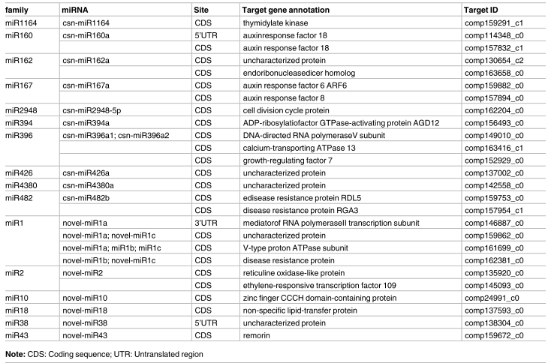
为了进一步预测和鉴定miRNA靶向基因,研究团队构建了降解组文库并进行深度测序,结合转录组数据和上述获得的miRNAs,通过TargetFinder软件预测到216个靶向基因被97个miRNAs调控。在这些靶向基因中,降解组测序鉴定了26个基因的26个切割位点,被16个miRNA家族的19个miRNAs进行转录后调控,如表1。
表1.通过降解组测序鉴定茶树1005中miRNA靶向基因
- 基于5’-RLM-RACE技术对目标断裂位点的验证
研究人员采用5’-RLM-RACE的方法,对其中5个有注释的miRNA(来自降解组测序)切割位点进行验证。结果显示,目标基因comp152929被csn-miR396a1 和 csn-miR396a2共同调控,切割位点在目标基因的第11-12个核苷酸处;两个基因comp159882和comp157894同时被miRNA167a调控,且有共同的切割位点,在目标基因的第11-12个核苷酸处;另外,miRNA394a调控comp156493,切割位点有两处,分别为第10-11,17-18个核苷酸处;同样的,novel-miR2调控comp145093,切割位点也有两处,分别为第10-11,12-13个核苷酸处。5’-RLM-RACE的实验验证了这些降解组分析的结果,另外一些切割位点有待进一步的验证。
7.茶叶中miRNA的表达模式分析
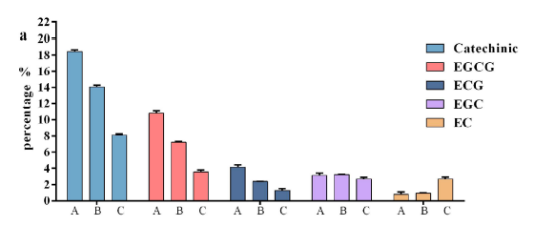
对茶树1005叶片儿茶素含量的研究发现,儿茶素在不同叶片中的的含量由高到低排序为:第一叶,芽,第二叶,第三叶,成熟叶。第一叶,第三叶和成熟叶的儿茶素含量有显著差异。
为了探索茶叶中miRNAs与儿茶素合成相关性,研究人员采用qRT-PCR的方法分析第一叶、第三叶以及成熟叶中miRNA(read values高于平均值)表达。通过表达模式的成簇分析将这些miRNA分成三组。第一组,miRNA表达模式与儿茶素含量成正相关,分别有novel-miR10, novel-miR13, novel-miR19, csn-miR165a, csn-miR170, csn-miR2593e 和novel-miR12;第二组,miRNA表达模式与儿茶素含量成负相关,分别有novel-miR1, novel-miR2, csn-miR160a, csn-miR162a, csn-miR394 和csn-miR396a;第三组,miRNA表达模式与儿茶素含量没有显著相关性,有csn-miR4380a。
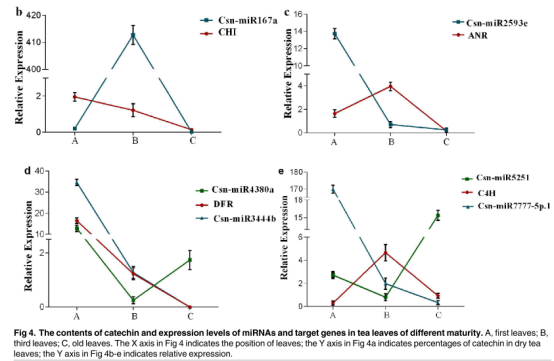

8.调控儿茶素生物合成基因miRNA的鉴定
为了深入了解miRNA在调控儿茶素合成代谢中的功能,研究人员将高通量测序获得的miRNA跟NCBI数据库以及RNA测序的儿茶素生物合成相关基因的mRNA进行BLAST,预测可能的miRNA。为了消除假阳性,对预测的断裂位点进行实验验证。通过5’-RLM-RACE实验验证了6个可能参与儿茶素生物合成功能基因表达调控的miRNA。它们分别是csn-miRNA167a、f csn-miR2593e、csn-miR4380a 、csn-miR3444b 、csn-miR5251 和csn-miR7777-5p.1。

9.儿茶素生物合成路径基因和miRNAs表达分析
为了更好的了解儿茶素生物合成过程中miRNA与目标基因之间的表达模式以及相互作用,研究人员将不同叶片中儿茶素生物合成相关基因和miRNA表达量进行分析,并且与儿茶素含量变化的模式相比较。结果显示,ANR和C4H有相似的表达模式,ANR在第三叶中表达量最高,它与EC,EGC和儿茶素含量没有明显的关系,在绿茶Shuchazao中有过类似的报道。CHI和DFR的表达量与儿茶素的含量成正相关的趋势,这与之前的报道一致。经分析发现,大多数的miRNA表达模式与目标基因呈相反的趋势。总之,miRNA和目标mRNA之间的表达模式和断裂位点的负相关性是儿茶素生物合成中目标miRNA靶向调控mRNA的有力证据。
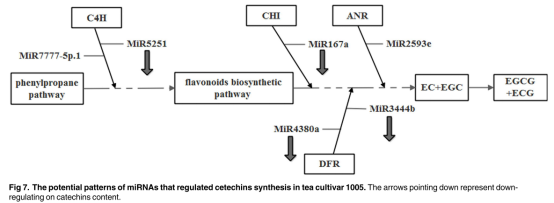
结论
该研究共鉴定了69个保守的miRNAs和47个新的miRNA。再结合降解组测序的和生物信息分析的方法预测了受19个miRNA调控的16个基因切割位点。通过5’-RLM-RACE实验对其中5个有注释的miRNA切割位点进行了验证。qRT-PCR结果显示:novel-miR1, novel-miR2,csn-miR160a, csn-miR162a, csn-miR394 和 csn-miR396a与儿茶素含量负相关。6个miRNA(csn-miRNA167a,csn-miR2593e,csn-miR4380a,csn-miR3444b,csn-miR5251和csn-miR7777-5p.1)及其与儿茶素生物合成相关的靶基因的表达也通过qRT-PCR进行了分析;这些miRNA和儿茶素含量之间呈正相关和负相关,而在其目标基因和儿茶素含量之间呈正相关。这个结果表明这些miRNA可能通过下调它们来负调节儿茶素生物合成生物合成相关靶基因。综上所述,miRNA是茶叶中关键的调节因子,5′-RLM-RACE和表达分析的结果揭示了miRNAs在儿茶素合成代谢中的重要作用。
创新点
本研究采用多种生物技术手段的联合分析(高通量测序,降解组测序,qRT-PCR技术和5’-RLM-RACE技术),寻找到新的调节儿茶素生物合成的miRNA,并且验证了miRNA调控的切割位点。