05月28 定位+实验验证:Plant Journal | BSA助力黄瓜耐水淹定位解析
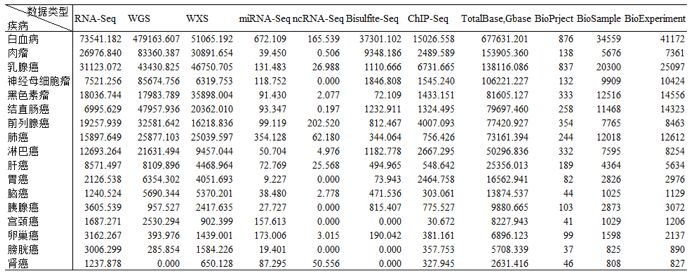
Bulked Segregant Analysis(BSA)分析是利用极端性状个体混池快速进行功能基因挖掘的常用方法,广泛应用在植物基因克隆方面。由百迈客和扬州大学陈学好教授课题组合作,利用SLAF-BSA策略定位黄瓜耐淹基因,相关研究成果发表于Plant Journal。 英文标题:The major-effect QTL CsARN6.1 encodes an AAA-ATPase domain-containing protein that is associated with waterlogging stress tolerance through promoting adventitious root formation 文献PDF全文免费下载: http://tour.biocloud.net/article/v1/into/articleDetail/29315927tour.biocloud.net 中文标题:CsARN6.1编码的AAA-ATPase基因通过促进不定根形成增强黄瓜耐水淹性 发表期刊:the Plant Journal, 2018 影响因子:5.90 实验流程 实验过程 1.不定根数目是数量性状且与水淹胁迫耐受力显著相关 表型观察发现,水淹处理7天后,Zaoer-N幼苗下胚轴生长出许多不定根,而Pepino几乎没有;通过对F2群体表型统计,所有子代的不定根数目表现出正态分布,这也说明了该性状为数量性状。另外,对F2群体的949个个体进行水淹耐受力评估打分,发现不定根数目与水淹耐受力之间呈显著正相关,皮尔森相关系数为0.72(P = 0.05),这表明不定根数目可以作为衡量水淹耐受力的可靠指标。 2、ARN6.1的初定位 利用SLAF-seq的方法对亲本及两个极端混池进行测序,亲本测序深度分别为29.18×和22.85×,两个混池的深度分别为50.6×和53.72×,以9930为参考基因组,利用△SNP-index和ED的方法计算显著关联位点,将关联区域定位在6号染色体标记SLAF_marker_192310和SLAF_marker_192096之间,区间大小301kb(图1)。 图1 BSA定位结果 3、ARN6.1的精细定位 利用定位区间侧翼SLAF标记(SLAF_marker_192310和SLAF_marker_192096)上的SNP,分别各开发KASP标记(KASP1和KASP13),并对2274个F2子代进行分析,结合基因分型和表型数据,将ARN6.1定位到61.5kb的区间(KASP10和KASP11)。为进一步缩小区间,利用KASP10和KASP11对4417个F2个体进行分型,得到6个重组个体,这6个重组个体分别自交得到6个F2:3家系,然后利用新开发的5个dCAPS对F2:3家系进行分型,结合所有表型数据,最终将ARN6.1定位在36.1kb的范围内。对该区进行注释,共有7个基因,有趣的是,其中5个基因都被预测为编码AAA 型的ATP酶家族蛋白。 2个亲本重测序分析,在36.1kb的区间内开发到25个SNP,研究者对100个黄瓜自交系的23个SNP(2个SNP只存在于Zaoer-N中而被过滤掉)进行分型,结合每个自交系不定根数目的表型数据进行局部关联分析,结果显示SNP02与表型有较强的关联性。对SNP02分析发现,其位于Csa6G504460的第二外显子,可能就是引起变异的SNP位点。 4、表达分析验证Csa6G504460 前期研究中,研究者对亲本Zaoer-N和Pepino幼苗下胚轴在水淹处理后进行转录组分析,以上定位区间内的7个基因只有Csa6G504460在处理组和对照组间存在差异表达,并且差异表达只发生在Zaoer-N中。而后,研究者对这7个基因又进行qRT-PCR分析,同样发现只有Csa6G504460在Zaoer-N的处理组和对照组间存在差异表达,并且在处理后36h表达量差异出现峰值。另外,组织特异表达分析表明Csa6G504460在多个组织中均有表达,但是在根中的表达量显著高于其他组织。因此,从基因表达角度验证了Csa6G504460(以下命名为CsARN6.1)的真实性。 5、CsARN6.1突变体降低ATP酶活性 基因组和cDNA序列分析显示,CsARN6.1拥有2个外显子,被预测为编码含有511个氨基酸残基的AAA-ATPase结构域蛋白,该蛋白中含有一个coiled-coil结构域(图2),前期关联到的SNP02即位于该结构域,由于该SNP的突变导致Asp被替换成Gly,Zaoer-N为CsARN6.1^Asp型,表现出较强的ATP酶活性,而Pepino为CsARN6.1^Gly型,几乎没有ATP活性。 图2 AAA-ATPase基因结构 6、转基因验证 为验证CsARN6.1的功能,将CsARN6.1^Asp通过PCR实验克隆后,转入拟南芥中,发现转基因植株的根长显著长于对照,同时,转基因植株上可以明显观察到侧根发育,而对照组则没有侧根发育。为进一步验证,研究者将CsARN6.1^Asp转入黄瓜品种Xintaimici(CsARN6.1^Gly型),并经多代自交和筛选,获得单拷贝的纯合转基因植株。发芽后3天,转基因植株的初生根长度显著长于野生型。水淹处理后,转基因黄瓜下胚轴中CsARN6.1的表达量显著高于野生型。处理后7天,转基因黄瓜下胚轴的不定根数目明显高于野生型。另外,野生型黄瓜叶片和子叶的萎黄病较转基因黄瓜严重。以上转基因结果证实CsARN6.1能够促进不定根形成和黄瓜水淹耐受力。 7、ATP酶活性影响黄瓜不定根形成 前期研究发现,EDTA能够抑制AAA-ATPase蛋白的ATP酶活性。研究者通过体外实验发现,经EDTA处理的CsARN6.1^Asp蛋白的ATP酶活性相对于对照降低24%(图3 a)。随后,研究者用加入EDTA的水处Zaoer-N幼苗,以检测ATP酶活性损耗对下胚轴不定根的影响。结果发现,经EDTA处理后,Zaoer-N没有了不定根生成能力(图3 b.c)。 进化树分析显示,CsARN6.1与拟南芥At2G18190和At3G50930存在较高的同源性(图3d),而在之前研究中发现,H2O2处理拟南芥后,At2G18190.1和At3G50930.1被显著诱导表达。水淹后植物体内H2O2积累是普遍的生理响应。因而研究者尝试在水中加入H2O2后处理Zaoer-N幼苗,与无水处理的对照相比,48h后处理组植株CsARN6.1的表达量是对照组的4.3倍,与不加H2O2的水处理的对照相比,48h后CsARN6.1的表达量是对照组的2.1倍(图3e)。5天后统计不同处理的材料下胚轴不定根数目,发现与不加H2O2的水处理的对照相比,水中加入H2O2的处理组的不定根数目增加60%(图3 f.g)。 图3 EDTA及H2O2处理对ATP酶和黄瓜生根的影响 总结 在该研究中,基因挖掘和功能分析使用了SLAF-seq、BSA分析、重测序、KASP、RNA-seq、qRT-PCR、PCR克隆等测序和分子实验方法 成功分离适应水淹的主效基因CsARN6.1,并验证基因功能,解析了黄瓜耐水淹分子机制。 百迈客云BSA分析工具是一款结合多年BSA分析项目分析经验开发的包含一键式标准化分析和个性化多样性分析集成式分析平台.可以进行基本分析和个性化分析,基本分析内容包括:1) 原始数据导入;2) 与参考基因组比对;3) BSA分析;4) 一键式生成网页版结题报告。 工具地址: https://international.biocloud.net/zh/software/agriculture/detail/6388C5BF964943B7B4B1DDF4811A1BD2international.biocloud.net BSA分析,即集群分离分析,它是通过具有相对性状的一对亲本杂交,在其任一分离后代群体中,根据个体表型(或基因型)的极端差异,选取一定量个体,将其DNA,RNA,SLAF-seq(百迈客自主研发的技术)混合,构建两个基因池(pool).然后将混池测序数据与参考基因组的序列比对,基于检测到SNP,InDel等变异类型,寻找两混池间存在显著差异标记,利用欧氏距离,SNP-index等算法评估与性状关联的区域.并对区域内的基因进行功能注释和富集分析等等.在基础上还可以进行深入挖掘,如:引物设计,区域内基因挖掘及标记筛选等等! 本文系“百迈客生物”发布,转载请联系作者。...